DESCRIPTION OF OUR BUSINESS
In this Business Section, unless the context indicates otherwise, references to “Biovest,” “the Company,” “our Company,” “we,” “us,” and similar references refer to Biovest International, Inc. and its subsidiaries, Biovest Europe, Ltd. and ViraCell Advanced Products, LLC.
Overview
Biovest is a biotechnology company focused on developing and commercializing BiovaxID™, as a personalized therapeutic cancer vaccine for the treatment of B-cell blood cancers; the continued development, commercialization, manufacture and sale of AutovaxID® and other instruments and disposables; and the commercial sale and production of cell culture products and services. We were incorporated in Minnesota in 1981, under the name Endotronics, Inc. In 1993, our name was changed to Cellex Biosciences, Inc. In 2001, we changed our corporate name to Biovest International, Inc. and changed our state of incorporation from Minnesota to Delaware.
As a result of our collaboration with the National Cancer Institute (“NCI”), we are developing BiovaxID as a personalized therapeutic cancer vaccine for the treatment of non-Hodgkin’s lymphoma (“NHL”), a B-cell cancer, specifically, follicular lymphoma (“FL”) and mantle cell lymphoma (“MCL”), and potentially other B-cell cancers. Both FL and MCL are generally considered to be incurable with currently approved therapies. These generally fatal diseases arise from the lymphoid tissue and are characterized by an uncontrolled proliferation and spread throughout the body of mature B-cells, which are a type of white blood cell. Three clinical trials conducted under our investigational new drug application (“IND”) have studied BiovaxID in NHL. These studies include a Phase 2 clinical trial and a Phase 3 clinical trial in patients with FL, as well as Phase 2 clinical trial in patients with in MCL. The Phase 3 randomized, controlled trial demonstrated a statistically significant disease free survival benefit in patients who received BiovaxID compared to patients who received control vaccine (46.0 vs. 30.6 months respectively, P= 0.029 adjusted; HR= 0.58, %95 CI: 0.360-0.960). We believe that these clinical trials demonstrate the safety and efficacy of BiovaxID.
Based on our scientific advice meetings with multiple European Union (“EU”)-Member national medicines agencies, on June 13, 2012 we filed our formal notice of intent to file a marketing authorization application (“MAA”) with the European Medicines Agency (“EMA”) which began the EU marketing approval application process. Subsequently, on December 3, 2013, we submitted an MAA with the EMA for BiovaxID. Additionally, based on a scientific advice meeting conducted with Health Canada, we announced plans to file a new drug submission application (“NDS”) seeking regulatory approval in Canada. We also conducted a formal guidance meeting with the U.S. Food and Drug Administration (“FDA”) in order to discuss the path for our filing of a biologics licensing application (“BLA”) for BiovaxID’s U.S. regulatory/marketing approval. As a result of this guidance meeting, we plan to conduct a second Phase 3 clinical trial to complete the clinical data gained through our first Phase 3 clinical trial and our BiovaxID development program to support our filing of our BLA for BiovaxID. We are preparing to initiate this second Phase 3 clinical trial, subject to required funding.
To support our planned commercialization of BiovaxID and to support the products of personalized medicine and particularly, patient specific oncology products, we developed and commercialized a fully automated, reusable instrument that employs a fully disposable, closed-system cell-growth chamber incorporating a hollow fiber cell-growth cartridge called AutovaxID. Since it is fully enclosed, computer controlled and automated, AutovaxID requires limited supervision and manpower to operate, compared to manual instruments. AutovaxID is suitable for growing antibody-secreting cell lines, including hybridomas and Chinese hamster ovary (“CHO”) cells, which are among the leading kinds of cell lines used for commercial therapeutic protein manufacture. AutovaxID has a small footprint and supports scalable production. We plan to utilize the AutovaxID technology to streamline the commercial manufacture of BiovaxID. We believe that AutovaxID is the first cell culture system that enables production of personalized cell-based treatments economically. AutovaxID uses a disposable production unit which provides for robust and dependable manufacturing while complying with the industry current good manufacturing practices (“cGMP”) standards. We are collaborating with the U.S. Department of Defense (“DoD”) and others to further develop AutovaxID and related hollow fiber systems and to explore potential production of additional vaccines, including vaccines for viral indications such as influenza and other contagious diseases. We also manufacture instruments and disposables used in the hollow fiber production of cell culture products. We manufacture mammalian cell culture products such as whole cells, recombinant and secreted proteins, and monoclonal antibodies for third parties, primarily researchers. We have produced over 7,000 cell based products for an estimated 2,500 researchers around the world. We consider our vast experience in manufacturing small batches of different cell based products, together with our expertise in designing and manufacturing instruments for personalized medicines as important competencies supporting our development of patient specific immunotherapies.
PRODUCTS
BIOVAXID™ - Personalized Therapeutic Cancer Vaccine
The Human Immune System
The immune system functions as the body’s natural defense mechanism for identifying and killing or eliminating disease-causing pathogens, such as bacteria, viruses, or other foreign microorganisms. However, with regard to cancer, including lymphomas, the immune system’s natural defense mechanism is believed to be largely thwarted by natural immune system mechanisms which seek to protect “self-cells” from attack. In humans, the primary disease fighting function of the immune system is carried out by white blood cells (leukocytes), which mediate two types of immune responses: innate immunity and adaptive immunity. Innate immunity refers to the broad first-line immune defense that recognizes and eliminates certain pathogens prior to the initiation of a more specific adaptive immune response. While the cells of the innate immune system provide a first line of defense, they cannot always eliminate or recognize infectious organisms. In some cases, new infections may not always be recognized or detected by the innate immune system. In these cases, the adaptive immune response has evolved to provide a highly-specific and versatile means of defense which also provides long-lasting protection (immune memory) against subsequent re-infection by the same pathogen. This adaptive immune response facilitates the use of preventative vaccines that protect against viral and bacterial infections such as measles, polio, diphtheria, and tetanus. We believe that BiovaxID creates an adaptive immune response to cancerous B-cells.
Adaptive immunity is mediated by a subset of white blood cells called lymphocytes, which are divided into two types: B-cells and T-cells. In the bloodstream, B-cells and T-cells recognize antigens, which are molecules that are capable of triggering a response in the immune system. Antigens are molecules from bacterial, viral, or fungal origin, foreign (non-self) proteins, and in some cases, tumor-derived proteins that can stimulate an immune response. The human body makes millions of different types of B-cells that circulate in the blood and lymphatic systems and perform immune surveillance. Each B-cell has a unique receptor protein (immunoglobulin) on its surface that binds to one particular antigen. Once a B-cell recognizes its specific antigen and receives additional signals from a T-helper cell, it can proliferate and become activated in order to secrete antibodies (immunoglobulins; Ig) which can neutralize the antigen and target it for destruction. T-cells may also recognize antigens on foreign cells, whereby they can promote the activation of other white blood cells or initiate destruction of the targeted cells directly. A person’s B-cells and T-cells can collectively recognize a wide variety of antigens, but each individual B-cell or T-cell will recognize only one specific antigen. Consequently, in each person’s bloodstream, only a relatively few lymphocytes will recognize the same antigen. Since B-cell cancers such as NHL are tumors arising from a single malignant transformed B-cell, the tumor cells in NHL maintain on their surface the original malignant B-cell’s immunoglobulin (collectively referred to as, the “tumor idiotype”) that is distinct from those found on normal B-cells. The tumor idiotype maintained on the surface of each B-cell lymphoma serves as the tumor-specific antigen for the BiovaxID cancer vaccine.
In many cases, including in NHL, cancer cells produce molecules known as tumor-associated antigens, which may or may not be present in normal cells but may be over-produced in cancer cells. T-cells and B-cells have receptors on their surfaces that enable them to recognize the tumor associated antigens. While cancer cells may naturally trigger a B- or T-cell-based immune response during the initial appearance of the disease, this response may be only weakly specific or attenuated in such a way that it does not fully eradicate all tumor cells. Subsequently, tumor cells gradually evolve and escape from this weak immune response and are able to grow into larger tumors. In addition, because cancer cells arise from normal tissue cells, they are often able to exploit or increase existing immune tolerance mechanisms to suppress the body’s immune response which would normally destroy them. In other cases, chemotherapy or other treatment regimens used to treat the cancer may themselves weaken the immune response and render it unable to reject and kill tumor cells. Even with an activated immune system; however, the number and size of tumors can often overwhelm the immune system.
In the case of cancer and other diseases, immunotherapies are designed to activate a person’s immune system in an attempt to combat the disease. There are two forms of immunotherapy used to treat diseases: passive and active. Passive immunotherapy is exemplified by the intravenous infusion into a patient of antibodies specific to the particular antigen. While passive immunotherapies have shown clinical benefits in some cancers, they require repeated infusions and can cause the destruction of normal cells in addition to cancer cells. An example of passive immunotherapy to treat lymphoma is monoclonal antibodies such as rituximab. An active immunotherapy, on the other hand, seeks to generate a durable adaptive immune response by introducing an antigen into a patient, often in combination with other components that can enhance an immune response to the antigen. BiovaxID is an example of active specific immunotherapy. Although active immunotherapies have been successful in preventing many infectious diseases, their ability to combat cancers of various types has been limited by a variety of factors, including the inability of tumor antigens to elicit an effective immune response, difficulty in identifying suitable target tumor antigens, inability to manufacture tumor antigens in sufficiently pure form, and inability to manufacture sufficient quantities of tumor antigens.
Nevertheless, in 2010 one active immunotherapy, Provenge® (sipuleucel-T) developed by Dendreon Corporation, received marketing approval from the FDA for the treatment of asymptomatic or minimally symptomatic metastatic castrate resistant (hormone refractory) prostate cancer. This represents the first case of an active immunotherapy to successfully gain marketing approval in the U.S. In March 2011, a second active immunotherapy, Yervoy® (ipilimumab), developed by Bristol-Myers Squibb received marketing approval from the FDA, for the treatment of late-stage metastatic melanoma. In addition to BiovaxID, there are a number of other active immunotherapeutics for cancer in various stages of clinical trials that have demonstrated promising results.
A number of features of the NHLs make these tumors particularly suitable for treatment with a therapeutic cancer vaccine. The malignant B-cell lymphocytes of NHL express a unique, identifiable tumor-specific antigen which is not expressed by other (healthy) cells in the body. In contrast, the majority of human cancers typically lack strong ubiquitous expression of tumor-specific antigens to distinguish them from normal cells, or they express a potentially widely-varying mix of antigens which can be difficult to identify and formulate into a successful therapeutic vaccine.
Non-Hodgkin’s Lymphoma
Non-Hodgkin’s lymphoma (“NHL”) is a heterogeneous group of malignancies of the lymphatic system with differing clinical behaviors and responses to treatment. BiovaxID™ has been studied in two distinct forms of NHL, namely, FL and MCL. NHL was the seventh most common type of cancer in the U.S. in 2011 (Lymphoma and Leukemia Society- Facts 2012), with an estimated prevalence of 484,336 cases in 2011 in the U.S. (Surveillance, Epidemiology, and End Results- SEER Stat Fact Sheets: Non-Hodgkin’s Lymphoma). NHL accounts for 3% of all cancer deaths in the U.S. (American Cancer Society- Facts and Figures 2012). NHL is one of the few malignancies in which there continues to be a rise in incidence. Since the early 1970’s, incidence rates for NHL have nearly doubled. Moreover, in spite of recent advances in the standard of care, the overall five-year survival rate remains at approximately 69% (Surveillance, Epidemiology, and End Results [SEER] Stat Fact Sheets: Non-Hodgkin Lymphoma, 2014). In 2012, it is estimated that 70,130 new cases of NHL will be diagnosed and 18,940 Americans will die from the disease (American Cancer Society- Facts and Figures 2012), with a comparable number estimated in Europe.
NHL is usually classified for clinical purposes as being either “indolent” or “aggressive,” depending on how quickly the cancer cells are likely to grow and spread. The indolent, or slow-growing, form of NHL has a very slow growth rate and may need little or no treatment for months or possibly years. The aggressive, or fast-growing, form of NHL tends to grow and spread quickly and cause severe symptoms, and patients with aggressive NHL have shorter overall survival (“OS”).
Follicular Lymphoma
Indolent (slow growing) and aggressive (fast growing) NHL each constitute approximately half of all newly diagnosed B-cell NHL, and roughly half of the indolent B-cell NHL is follicular lymphoma (“FL”). Accordingly, approximately 22% of new cases of NHL fall into the category of disease known as indolent FL, which translates in about 106,550 cases in 2011 in the U.S. (SEER Stat Fact Sheets: Non-Hodgkin’s Lymphoma). We have conducted a Phase 2 clinical trial followed by a Phase 3 clinical trial in FL under our IND. FL is a form of NHL that is derived from a type of cell known as a follicle center cell. Despite its slow progression, FL is almost invariably fatal. According to data from 2003-2009 from 18 SEER areas, the 5-year relative survival rate in FL patients is 85.4%. According to a recent publication based on data from patients treated at Stanford University during 1997-2003 (Tan et al., Blood 2013), after the introduction of rituximab OS in patients with grade 1-2 FL (i.e., indolent lymphoma) reached 72% at 10-years. This improvement in OS was attributed to better supportive care and effective therapies for relapsed disease. The current standard of care for treatment of advanced, bulky FL (bulky Stage II, Stage III-IV) as specified by the National Comprehensive Cancer Network (“NCCN”) includes initial treatment of newly-diagnosed patients with rituximab-containing chemotherapy. Rituximab is a monoclonal antibody (an immune protein capable of selectively recognizing and binding to a molecule) which targets a protein primarily found on the surface of both healthy and cancerous B-cells, known as CD20. Accordingly, rituximab seeks to bind and destroy all B-cells, including healthy B-cells, as a means of controlling the progression of FL in treated patients. Rituximab and other biologics currently approved for lymphoma are characterized as “passive immunotherapies”. Following administration, rituximab exerts its effects primarily through an unselective and near total destruction of a patient’s B-cells, including malignant as well as healthy B-cells. Rituximab and other passive immunotherapies are often administered in sequential, repeated doses to achieve their effect, and following cessation of administration are over time eliminated from the patient’s circulation by normal bodily functions. Rituximab is characterized as a targeted therapy since it targets CD20, which is present on both healthy and tumor cells. Rituximab is manufactured in bulk and is not considered to be a personalized therapy. By comparison, BiovaxID™ is characterized as an “active immunotherapy”. Active immunotherapies attempt to stimulate the patient’s immune system to respond to a disease. “Specific active immunotherapies” such as BiovaxID, specifically seek to induce cellular and/or humoral immune responses focused on specific antigens present on a diseased cell (such as a tumor cell). As a specific, active immunotherapy, BiovaxID targets only the cancerous B-cells while sparing healthy B-cells. Accordingly, BiovaxID is highly targeted. BiovaxID is manufactured specifically and entirely for each patient and is considered to be a highly “personalized therapy”. If approved, BiovaxID will represent the only specific active immunotherapeutic approved for the treatment of FL and therefore will represent a new class of drugs that provides a new therapeutic option for patients with lymphoma. In February 2011, the NCCN issued treatment guidelines recognizing “consolidation therapy” as a defined treatment category for FL in first remission. Consolidation therapy options differ from induction therapies in that they primarily seek to prolong first remission duration by consolidating the effects of induction therapy, which primarily seeks to reduce active, bulky tumor masses. The following anti-CD20 monoclonal antibody drug products are currently approved consolidation treatment options for the first-line consolidation or extended dosing in FL: Rituxan® and Zevalin®. All of these treatment options are passive immunotherapies that result in profound B-cell depletion. Following the results of the Eastern Cooperative Oncology Group E4402 protocol, also called RESORT (Rituximab Extended Schedule or Re-treatment Trial), reported at the 2011 annual meeting of the American Society of Hematology (“ASH”), the NCCN revised its clinical practice guidelines on FL. Since 2012 NCCN guidelines have considered Rituxan maintenance therapy and Zevalin consolidation therapy as ‘optional’ therapeutic approaches post-induction therapy, rather than ‘recommended’ therapeutic approaches.
Current U.S. Approved Consolidation Therapies for NHL and Urgent Need for Alternative Treatment Options
Rituxan® (rituximab): Rituximab maintenance consists of administration of the anti-CD20 antibody rituximab administered at a dose of 375 mg/m2 every 8 weeks for 24 months (12 injections) administered by IV infusion every 8 weeks starting 8 weeks ± 7 days after the last induction treatment (whether immuno-chemotherapy or rituximab, whichever is later). Administration of rituximab (and other anti-CD20 agent) maintenance extends the profound immunosuppression achieved by induction therapy, as it targets the pan-B-cell CD20 protein. This continued dosing of the induction agent induces profound B-cell depletion for the two-year duration of the regimen.
Zevalin® (ibritumomab tiuxetan): Zevalin is an immunoconjugate resulting from covalently-bonded anti-CD20 antibody ibritumomab and the linker-chelator tiuxetan [N-[2-bi(carboxymethyl)amino]-3-(p-isothiocyanatophenyl)-propyl]-[N-[2-bis(carboxymethylamino]-2-(methyl)-ethyl]glycine. This linker-chelator provides a high affinity, conformationally restricted chelation site for Indium-111 or Yttrium-90. Administration follows induction rituximab and requires preliminary dosimetry and imaging administration of In-111 (Day 1) followed by administration of Y-90 Zevalin on Day 7, 8, or 9. The maximum allowable dose of Y-90 Zevalin is 32.0 mCi (1184) MBq and physicians and patients receiving the agent must exercise radiation exposure precautions upon administering or handling the agent.
Urgent Need for New Consolidation Treatment Option for NHL: The currently approved consolidation agents are no longer recommended therapeutic options post-induction therapy by the NCCN clinical practice guidelines. Following the results of the Eastern Cooperative Oncology Group E4402 protocol, also called RESORT (Rituximab Extended Schedule or Re-treatment Trial), reported at the 2011 annual meeting of ASH, the 2012 NCCN revised guidelines include rituximab maintenance and radioimmunotherapy as ‘optional’ therapeutic options post-induction. BiovaxID™ is expected to offer a non-immunosuppressive alternative to rituximab maintenance as a consolidation therapy for FL and MCL. By its mechanism of action, BiovaxID is not riddled with the risk of development of rituximab-resistance. Thus, BiovaxID represents a potential novel option for consolidation therapy that is safe and effective, compatible with current induction regimens, and unlikely to interfere with future therapies while potentially increasing the utility of other therapies.
Mantle Cell Lymphoma
Mantle cell lymphoma (“MCL”) is a rare, aggressive subtype of NHL characterized by short remissions and rapid progression similar to aggressive lymphomas and successive relapses, reflecting incurability similar to indolent lymphomas. The median OS for MCL has been cited as 5-7 years (Perez-Galan et al., 2011). MCL represents approximately 6% of all NHL cases and worldwide there are approximately 7,800 new cases each year of which, approximately, one half are in the U.S. (see “Current treatment approaches for mantle-cell lymphoma” J Clin Oncol. Sep 10 2005 and “New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes.” J Clin Oncol. Aug 1998). The majority of MCL patients have disseminated disease and bone marrow involvement at diagnosis. Patients’ clinical outcomes from currently available therapies are poor. Although many therapeutic regimens are capable of rendering high initial response rates, these responses are of short duration (i.e., about 20 months) and the relative survival rates of MCL patients are among the lowest compared to other types of NHL. After a patient’s first relapse, the expected disease course and prognosis is very poor, with an expected median OS of about 1-2 years. No currently available therapeutic regimens are curative.
While there are several therapeutic regimens available to treat MCL patients, there currently exists no consensus standard of care for treatment of first-line relapsed MCL. As such, MCL remains incurable and it is generally considered that additional treatment options are required given this significant unmet medical need. Currently, upon first diagnosis MCL patients are often evaluated for eligibility for autologous stem cell transplantation (“autoSCT”). Stem cell transplantation, an aggressive treatment protocol consisting of high-dose chemotherapy, immunotherapy and full-body radiation, aims to treat the patient’s tumor and purge the bone marrow of lymphoma cells. MCL patients who are eligible for autoSCT receive either R-CHOP (rituximab, cyclophophamide, doxorubicin, vincristine, prednisone) followed by autoSCT or R-HyperCVAD (rituximab, cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with rituximab plus high dose methotrexate and cytarabine) followed by observation. Although these therapeutic approaches do yield high response rates, they are associated with high rates of adverse events and treatment discontinuation, high risk of myelodysplastic syndrome, and high mortality rates. Consequently, the considerable toxicity associated with these regimens largely limits these options primarily to a select subset of the MCL patients who are younger and better fit to tolerate these high-intensity treatments. However, even this subset ultimately gains only modest benefits from existing treatment options. Moreover, the use of these more aggressive regimens appears not to result in superior OS as compared to standard therapies. Given that the median age for newly diagnosed MCL patients is 60 years, less aggressive therapeutic approaches are needed.
Development Status of BiovaxID™
Preliminary studies demonstrated that treatment of patients with NHL with an active immunotherapy could allow a patient’s immune system generate clinically significant immune responses. These studies have been published in The New England Journal of Medicine (October 1992), Blood (May 1997), and Nature Medicine (October 1999). In the treatment of cancer, residual tumor cells remaining in the patient after completion of surgery or anti-tumor therapy are often the cause of tumor relapse. These residual tumor cells cannot always be detected by standard imaging techniques but their destruction may be feasible by active immunotherapy. The use of such vaccines differs from traditional cancer treatment in that the ultimate mechanism of action against the tumor is indirect: the anti-tumor immunity induced by vaccination, rather than the vaccine itself, is ultimately responsible for treatment benefit. In 1994, the NCI filed for initiation of an IND for the purpose of conducting clinical trial(s) investigating the use of BiovaxID in NHL. Under this IND and also in 1994, the NCI began the Phase 2 clinical trial in FL; in 1999, the Phase 3 clinical trial in FL; and in 2000 a Phase 2 clinical trial in MCL. The NCI selected us to produce BiovaxID for the initial Phase 2 clinical trial in FL. In 2001, we entered into a formal cooperative research and development agreement (“CRADA”) with the NCI which formalized our collaboration with the NCI. In April 2004, the IND filed by the NCI was formally transferred to us, which made our Company the exclusive sponsor of the IND with full rights to complete the NCI-initiated Phase 3 clinical trial in FL and the NCI-initiated Phase 2 clinical trial in MCL, to communicate and negotiate with the FDA relating to marketing approval for BiovaxID and to conduct other clinical studies in NHL under the IND.
BiovaxID™ Clinical Trials
Phase 2 Clinical Trial of BiovaxID™ for Treatment of FL
In 1994, the Phase 2 clinical trial (NCT00878488) was commenced by the NCI to evaluate the ability of BiovaxID to eradicate residual lymphoma cells in 20 patients with FL who were in chemotherapy-induced first clinical complete remission (“CR”). All 11 patients with a detectable lymphoma gene sequence (translocation) in their primary tumors had cells from the malignant clone detectable in their blood by DNA polymerase chain reaction (“PCR”) analysis both at diagnosis and after chemotherapy, despite being in CR. In this clinical trial, molecular remission was defined as patients lacking any detectable residual cancer cells bearing the translocation as determined by a very sensitive PCR technique. After vaccination, 8 of these 11 patients converted to lacking cells in their blood from the malignant lymphoma clone detectable by PCR. Anti-tumor T-cell responses were found in the vast majority of the patients (19 of 20 patients), whereas anti-tumor antibodies were detected, but apparently were not required for molecular remission. Vaccination was thus associated with clearance of residual tumor cells from the blood and long-term disease-free survival. The demonstration of molecular remissions besides uniform, specific T-cell responses against lymphoma tumor targets, as well as the addition of granulocyte-monocyte colony-stimulating factor (“GM-CSF”) to the vaccine formulation provided the rationale for the initiation of a larger Phase 3 clinical trial at the NCI in 2000. These results were published in Nature Medicine (Bendandi et al., 1999). At the latest data follow-up after a median of 167.9 months (range: 10.0- 214.3 months) median DFS was 62.5 months (Figure 2), median OS was not reached, and 80% of the patients were still alive.
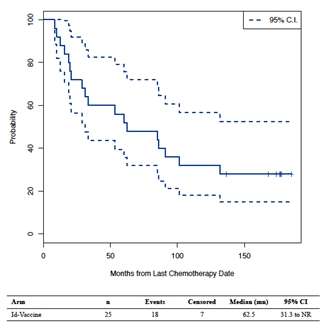
Figure 1: Disease free survival in the Phase 2 clinical trial in FL patients
Phase 2 Clinical Trial of BiovaxID™ for Treatment of MCL
In 2000, the NCI initiated a Phase 2 open-label clinical trial (NCT00020215) of BiovaxID for the treatment of MCL. This Phase 2 clinical trial was based upon the NCI’s Phase 2 clinical trial in FL. The primary objective of this Phase 2 clinical trial was to study BiovaxID in treatment-naïve patients with MCL and to determine the safety and efficacy of BiovaxID following a rituximab-based immunotherapy. Twenty-six patients with untreated, mostly (92%) stage IV MCL, were enrolled. All patients received 6 cycles of EPOCH-R (which is a chemo-immunotherapy consisting of etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab); 92% of the patients achieved CR and 8% achieved partial response (“PR”). All but 3 patients (i.e., due to disease progression or inability to manufacture the heterohybridoma vaccine) received BiovaxID together with keyhole limpet hemocyanin (“KLH”), an immunogenic carrier protein, on day 1, along with GM-CSF (100 µg/m2/day) on days 1-4 at 1, 2, 3, 4, and 6 months starting at least 3 months post-chemotherapy.
The results of our MCL Phase 2 clinical trial were reported in Nature Medicine (Neelapu et a., 2005). As reported in Nature Medicine, at 46 months of follow-up the OS was 89%, the median event-free survival (“EFS”) was 22 months, and 5 patients remained in continuous first CR. Antibody responses to immunization were detected in 30% of the patients, following a delayed pattern (i.e., detected mostly after the 4-5th vaccination) which paralleled the peripheral blood B-cell recovery. Most importantly, specific CD4+ and CD8+ T-cell responses were detected in 87% of patients post-vaccine, and in 7 of 9 patients tested these responses were detected after the 3rd vaccination when peripheral B-cells were by and large undetectable. The detected cytokine release response included GM-CSF, INF-g, and TNF-a (type I). In this study, BiovaxID induced both humoral and cellular immune responses following almost complete depletion of B-cells following rituximab-containing chemotherapy. The adverse events observed in this clinical trial were minimal and were limited mostly to injection site reactions. The results of the latest follow-up of these patients performed in 2011 were presented at the 2011 annual meeting of ASH (Grant et al., ASH 2011 Abstract #2707). At a median follow-up of 99 months (range, 15.2 - 131.0 months), median PFS was 24.1 months, median OS was 99.04 months, and median TTNT was 37.8 months. This median PFS of 24.1 months is longer than the median PFS for combination chemotherapy (16 - 17 months) (Dreyling et al, 2005; Mangel et al, 2004) or CHOP-R chemotherapy (21 months) (Lenz et al, 2005). Similarly, the median OS of 99.04 months greatly exceeds the 5- 6 years obtained with current therapies (Dreyling & Hiddemann, 2009), and raises the possibility that BiovaxID vaccination modified the natural history of MCL.
The association between OS and TTNT and tumor-specific GM-CSF cytokine induction observed at this long-term follow-up and presented at the 2011 annual meeting of the ASH and at the 2012 annual meeting of the American Society of Clinical Oncology (“ASCO”) suggests that the mechanism of action of BiovaxID is T-cell mediated and not B-cell (humoral) mediated. Patients with pre-vaccine-adjusted increases in tumor-specific normalized GM-CSF cytokine induction above the median value of 4 had a significantly longer median OS (not reached vs. 78.72 months, P= 0.006; Figure 2) and longer median TTNT (68.89 vs. 27.83 months, P = 0.026; Figure 4) compared to patients with pre-vaccine-adjusted increases in tumor-specific normalized GM-CSF cytokine induction < 4. There was no association between OS and specific anti-Id B-cell (humoral) responses or any other type of specific cellular responses. Additionally, the high impact of GM-CSF cytokine response mediated by antitumor T-cells on OS and TTNT suggest that anti-tumor immune responses significantly delayed tumor growth. In conclusion, these results demonstrate that administering BiovaxID post-rituximab is justifiable, as the effectively induced cellular responses underlie the fundamental mechanism of action of cancer vaccines. Given that rituximab-chemotherapy induction regimens represent the current therapeutic approach in B-cell lymphoma patients, BiovaxID is a compatible, effective, and safe consolidation therapy option.
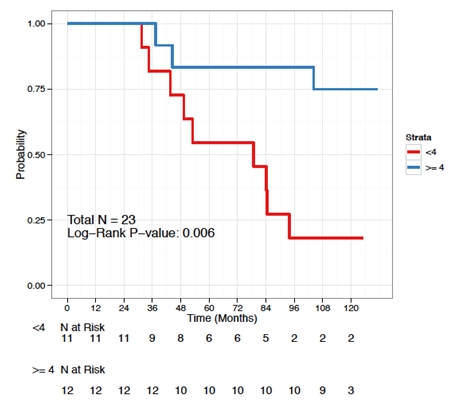
Figure 2. Overall Survival by GM-CSF Cytokine Response (< and > than median)
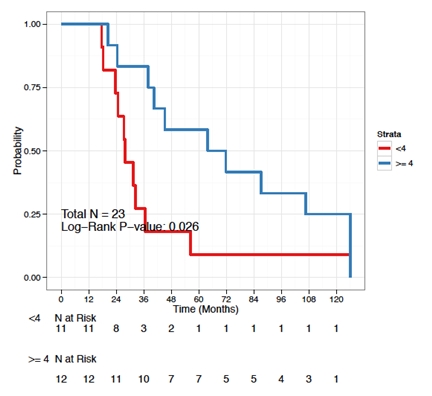
Figure 3. Time to next treatment by GM-CSF Cytokine Response (< and > than median)
Phase 3 Clinical Trial of BiovaxID™ for Treatment of FL
Overview and Objectives. In January 2000, the Phase 3 clinical trial in FL (NCT00091676) was initiated by the NCI. The Phase 3 clinical trial was a multi-center, double-blind, randomized, controlled clinical trial that was designed to confirm the results reported in the NCI’s Phase 2 clinical trial.
As studied in the Phase 3 clinical trial, BiovaxID consisted of the patient-specific idiotype protein (Id) derived from the patient’s cancer cells conjugated or combined with KLH and administered with GM-CSF which is a biological response enhancer. The comparator studied in the Phase 3 clinical trial was a control vaccine consisting of KLH and administered with GM-CSF. Accordingly, the only difference between BiovaxID and the control vaccine was the inclusion of the idiotype protein from the patient’s own tumor in BiovaxID. BiovaxID or the control vaccine was administered following chemotherapy (also referred to as induction therapy) with a drug combination, of prednisone, doxorubicin, cyclophosphamide, etoposide (referred to as “PACE”). Induction therapy represents the “first-line” treatment for FL patients and attempts to induce complete tumor remission as defined by radiological evidence (“CT scans”). In FL, patients treated with the current standard of care often achieve complete remission but these remissions almost always are of limited duration and most treated patients must eventually be re-treated for their disease. In the majority of cases, however, even with re-treatment, the disease often relapses and develops resistance to therapy, leading to a need for bone marrow transplant and eventually resulting in the death of the patient. In the Phase 3 clinical trial, patients who achieved complete response following induction therapy were assigned to a limited waiting period prior to vaccination to allow for immune reconstitution following the induction chemotherapy. Patients who relapsed during this immune reconstitution period did not receive either BiovaxID or control treatment. Patients who maintained their complete remission following this immune recovery period received either BiovaxID or control administered as 5 subcutaneous injections monthly over a six month period (one month was skipped).
The primary objective of the Phase 3 clinical trial was to definitively confirm the safety and efficacy of BiovaxID (autologous immunoglobulin follicular lymphoma idiotype vaccine) assessed by significant prolongation of clinical DFS following BiovaxID immunization with GM-CSF in FL patients achieving a CR/CRu with standard dose chemotherapy, when compared to DFS following administration of KLH-KLH and GM-CSF.
The secondary objectives of the Phase 3 clinical trial included:
(1) to determine the ability of BiovaxID to produce a molecular CR in subjects in clinical CR, but with PCR evidence of residual disease after standard chemotherapy;
(2) to determine the impact of BiovaxID on molecular remission in FL patients;
(3) to evaluate the ability of BiovaxID to generate an immune response against autologous tumor;
(4) to determine and compare the OS of subjects randomized to receive either treatment assignment; and
(5) to evaluate the safety of BiovaxID administered with GM-CSF.
Biopsy, Chemotherapy, and Immune Recovery. Prior to chemotherapy, a small tumor biopsy was performed to obtain tissue for tumor classification and characterization, and to provide starting material necessary to manufacture BiovaxID. Following this biopsy patients were initially treated with PACE chemotherapy in order to induce a CR or a complete response unconfirmed (“CRu”) as measured by CT scans. The clinical trial protocol stipulated that for all patients, an immune recovery period of approximately 6 months following completion of chemotherapy was required to be completed without relapse (“Immune Recovery Period”) before vaccination. The Immune Recovery Period was required in order to maximize the potential for immune response to vaccine and to avoid confounding factors from any potential lingering immunosuppressive effects of chemotherapy.
Randomization to Immune Recovery Followed by BiovaxID or Control. When the NCI designed the Phase 3 clinical trial in FL protocol, a decision was made to randomize patients, immediately after completion of chemotherapy and not to wait for the completion of the Immune Recovery Period in an effort to avoid expending NCI resources to manufacture patient-specific vaccines for patients who were not anticipated to receive the vaccine (e.g., control patients). In the Phase 3 clinical trial, of the 234 patients initially enrolled into the clinical trial, 177 patients completed chemotherapy, achieved CR/ CRu, and were randomized. As per the design of the Phase 3 clinical trial, patients who relapsed during the Immune Recovery Period were excluded from treatment with BiovaxID or control notwithstanding the fact that they had been randomized. In the Phase 3 clinical trial, of the 177 initially randomized patients, 117 remained eligible to be treated with either BiovaxID (76 patients) or control (41 patients) at the end of the Immune Recovery Period. Sixty patients of the 177 randomized patients were not treated with either BiovaxID or control mostly due to relapse during the immune recovery period (see Figure 4).
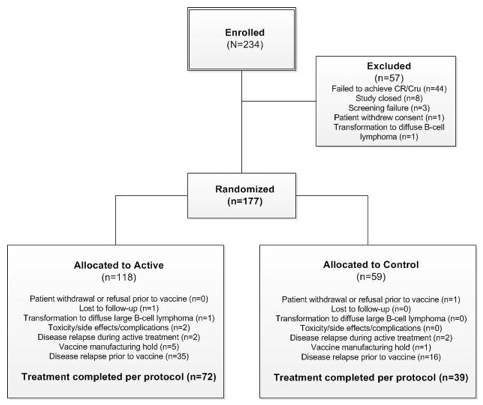
Figure 4. Enrollment, randomization, and treatment, Two hundred thirty-four patients were enrolled, and 177 patients were randomized to receive at least one dose of the blinded vaccine; 76 patients received Id-vaccine and 41 received control vaccine.
Trial Enrollment and the Use of Rituximab-Containing Induction Chemotherapy. During the course of the Phase 3 clinical trial, the standard of care for induction chemotherapy in FL changed to include rituximab, which reduced the ability to recruit and enroll patients into the study. In order to facilitate enrollment in the clinical trial, we amended the study protocol in 2007 to permit the use of a rituximab-containing chemotherapy regimen (“CHOP-R”), as induction therapy. However, the FDA requested that we abstain from vaccinating any patients who received CHOP-R and we did not vaccinate any of the patients who received CHOP-R chemotherapy under the Phase 3 clinical trial protocol.
Due to the protracted enrollment, the Phase 3 clinical trial’s Independent Data Monitoring Committee (“DMC”; a committee responsible for reviewing the available unblinded clinical trial data in the study and responsible for recommendations to the sponsor and the FDA) recommended an interim analysis of the clinical trial’s endpoints and overall safety profile which resulted in the termination and halting of the trial in 2008. As of April 15, 2008, when the Phase 3 clinical trial was officially closed, a total of 234 subjects had been enrolled and 177 subjects had been randomized. The total number of subject was less than the original planned sample size which called for 629 subjects to be enrolled and 540 to be randomized. While the termination of the Phase 3 clinical trial before completion of the planned accrual resulted in a smaller sample size than was originally intended, we believe that the randomized nature of our Phase 3 clinical trial yields a valid conclusion because the baseline characteristics of the patients in the 2 groups were balanced, the allocation to treatment arms was concealed, and the study was double-blinded.
Results of Phase 3 Clinical Trial. Of the 177 randomized patients in the ITT (intent to treat) population, 60 were censored due to ineligibility post-randomization due to the following: pre-vaccine relapse (n = 51, 28.8%), vaccine manufacturing hold (n = 6, 3.4%), and 1 each (0.6%) for lost to follow-up, withdrawal prior to vaccination, and transformation to diffuse large B-cell lymphoma. Importantly, relapse events that disqualified 51 patients for post-randomization treatment occurred in a blinded manner and were independent of external factors and were a consequence of each patient’s disease. Post-randomization relapse was equivalent between the two treatment groups: 29.7% versus 27.1%, active (BiovaxID+GM-CSF) to control (KLH-KLH+GM-CSF), respectively. Due to vaccine manufacturing failure, 5 patients who were randomized to receive active treatment (i.e., BiovaxID+GM-CSF) actually received control treatment (i.e., KLH-KLH+GM-CSF) but were analyzed as randomized, as per the prospective protocol provisions. In the ITT population there were no statistically significant differences in demographics and baseline characteristics between the treatment arms.
At a median follow-up of 56.6 months (range: 4.2 – 92.0 months), median DFS in the ITT population was 46.0 months for patients in the active treatment group (BiovaxID+GM-CSF) versus 30.6 months for patients in the control treatment group (KLH-KLH+GM-CSF) (log rank P = 0.029, controlled for FLIPI risk group and number of chemotherapy cycles) (Figure 5). BiovaxID vaccination therefore resulted in a 42% decrease in risk of relapse compared to control vaccination (HR= 0.58; %95CI: 0.370- 0.960).Median OS was not reached in either arm.
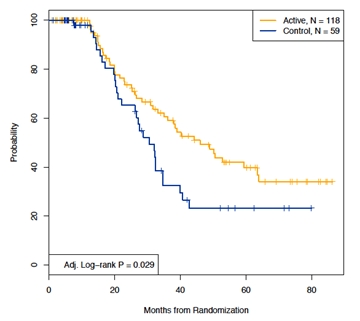
Figure 5. Disease-free survival (DFS) according to treatment arm for ITT Population (N = 177)
Analysis of Patients by Isotype. A typical antibody (“immunoglobulin”), including the lymphoma immunoglobulin expressed on the surface of each cancerous lymphoma cell, is composed of protein “heavy chains” and “light chains”. In humans, the heavy chains are classified as IgG, IgM, IgA, IgD and IgE, and the light chains are classified as either kappa or lambda. The Id protein expressed on the surface of FL cells is characteristic of the single B-cell from which the tumor arose. The immunoglobulin protein contains a region known as the “heavy chain” and a region known as the “light chain” (see Figure 6). Almost always in FL, the heavy chain region is characterized as either an IgM-isotype or an IgG-isotype. Figure 6 illustrates the dramatic differences in the structure of immunoglobulin protein characterized as an IgM-isotype as opposed that characterized as an IgG-isotype. Accordingly, an antibody may be referred to as IgG-isotype or IgM-isotype depending on its heavy-chain classification. In the normal immune response, antibody isotypes may have different roles and may help direct the appropriate immune response. The small region at the tip of the antibody is known as the “variable region”, or antibody binding site, and the balance of the isotype is known as the “constant region”. When we manufacture BiovaxID, we screen each patient’s tumor cells obtained by biopsy for the isotype. Approximately, 60% of patients with FL are diagnosed with tumors expressing an IgM isotype and approximately 40% of patients bear tumors expressing an IgG isotype. In rare cases (<1%), patients are diagnosed with another isotype (e.g. IgA). Infrequently, the patient’s tumor also contains cells with one or more isotype (a heterogenous or “mixed” isotype); in these patients we select either an IgG or IgM isotype for manufacture of BiovaxID. Each patient’s tumor isotype can be readily determined by standard analytical techniques (flow cytometry) at the time of the patient’s tumor biopsy. In both the Phase 2 and Phase 3 clinical trials in FL patients, the determination of tumor heavy-chain isotype determined the specific manufacturing and purification process used to make that patient’s vaccine. For patients who have tumors expressing an IgG (or an IgG-containing “mixed” isotype), we manufacture an IgG isotype vaccine and for patients determined to have tumors expressing an IgM (or an IgM-containing “mixed” isotype), we manufacture an IgM vaccine. Due to our manufacturing process (rescue fusion hybridoma), the isotype (IgG or IgM) of the tumor is entirely reproduced in each patient’s vaccine so that each patient’s BiovaxID vaccine matches the patient’s original tumor isotype (IgG or IgM).
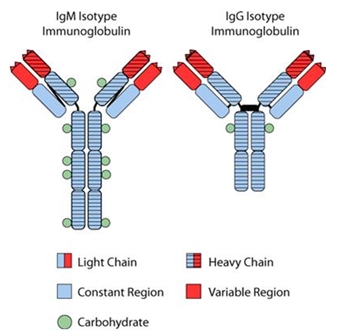
Figure 6. The Id protein expressed on the surface of FL cells is an immunoglobulin protein characteristic of the single B-cell from which the tumor arose.
Preclinical data indicates that the ability to develop an immune response differs between IgM-isotype and IgG-isotype idiotypes (De Groot, 2008; Reitan, 1995; Reitan, 2002); however, we do not currently have immune response data from human clinical trials to confirm this preclinical data. The IgG-isotype idiotype was reported to be tolerogenic, meaning that the immune response against the specific tumor target is suppressed. On the other hand, the IgM-isotype idiotype was reported to be highly immunogenic, meaning that it induces an ample, persistent immune response against the specific tumor target. The unique feature of our Phase 3 clinical trial was the manufacturing and administration of tumor-matched isotype idiotype vaccines which, allowed us to investigate whether these preclinical data translate into differential clinical efficacy of the two isotype vaccines in our clinical trial.
In an unplanned subgroup analyses, DFS was analyzed by tumor Ig heavy chain isotypes (IgM and IgG). Five patients from the ITT population were excluded from these analyses because their tumors isotype was mixed IgM/ or IgD. The IgM subgroup included 94 patients (N= 61 active, N= 33 control), and the IgG subgroup included 78 patients (N= 55 active, N= 23 control). In both isotype subgroups there were no statistically significant differences in baseline patient characteristics between experimental and control arms. Among patients receiving an IgM-Id vaccine, median time to relapse after randomization was 52.9 months versus 28.7 months in IgM tumor isotype control-treated patients (P= 0.004; HR= 0.33; 95% CI: 0.157- 0.727) (Figure 7). Among patients receiving IgG-Id vaccine, median time to relapse after randomization was 36.2 months versus 32.4 months in IgG tumor isotype control-treated patients (P= 0.703; HR= 1.19; 95% CI: 0.472- 3.036) (Figure 8). Cox proportional hazard modeling including the main effects of treatment arm and tumor isotype,, adjusted for FLIPI risk group and the number of chemotherapy cycles received, supports a differential effect of treatment by isotype as indicated by the P value of the interaction term between treatment and tumor Ig isotype (P= 0.062).
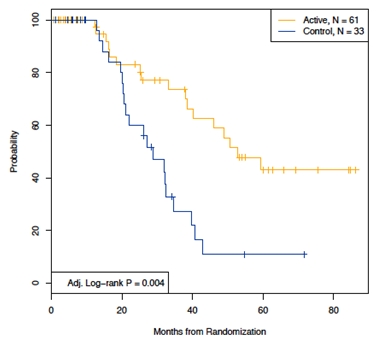
Figure 7. Disease free survival in IgM isotype patients
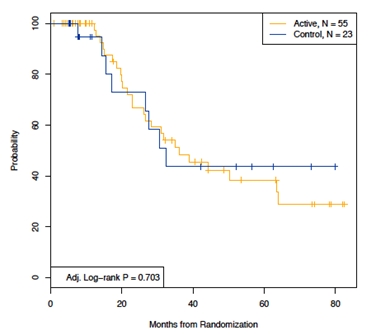
Figure 8. Disease free survival in IgG isotype patients
Our Phase 3 clinical trial had two unique manufacturing features, where: (a) the vaccine consists of the full structure of the idiotype protein (that is, both the variable and the constant regions of the immunoglobulin) and (b) the idiotype of the vaccine matches the idiotype of the patient’s own tumor. These unique features allowed us to be the first to investigate the clinical efficacy implications of the two tumor isotypes. The prior Phase 3 clinical trials of FL idiotype vaccines conducted by Genitope Corporation (“Genitope”) and Favrille, Inc. (“Favrille”) used a manufacturing process known as recombinant manufacturing that universally linked the patient’s variable region of the idiotype into an IgG isotype without regard to the actual isotype of each patient’s tumor. We believe that the use of an IgG isotype was due to the comparative ease of manufacture and purification of IgG proteins as well as to their relatively long half-life. There are two implications of the manufacturing processes used by these prior clinical trials: (1) clinical efficacy cannot be compared by isotype group; and (2) the lack of clinical efficacy observed in these clinical trials may be due to the tolerogenic effect of the universal IgG isotype used in the vaccine manufacturing. As such, we believe that our analysis by tumor isotype may provide profound insight into the efficacy of BiovaxID and may also suggest methods by which cancer vaccines in general could be developed in the future.
BiovaxID Regulatory and Marketing Status
Under our IND for BiovaxID, two Phase 2 clinical trials and one Phase 3 clinical trial have been completed studying BiovaxID for the indications of FL and MCL. We believe that these clinical trials demonstrate the safety and efficacy of BiovaxID and we have presented comprehensive summaries of the clinical development and clinical data to regulatory authorities in the EU, Canada and the U.S.
Based on our scientific advice meetings with multiple EU-Member national medicines agencies, on June 13, 2012, we filed our formal notice of intent to file a MAA with the EMA, which began the EU marketing approval application process. In response to our notice of intent to file for marketing approval, the EMA notified us that we were eligible to submit our planned MAA for BiovaxID under the EMA’s centralized procedure, as an orphan medicinal product for the treatment of FL. Under the EMA centralized procedure, the marketing approval of BiovaxID can be simultaneously obtained throughout all EU-member countries with a single MAA. Also, as part of the EMA’s centralized procedure, our MAA for BiovaxID will be assessed by the EMA’s Committee for Medicinal Products for Human Use (“CHMP”), which designates from within its membership, a Rapporteur and Co-Rapporteur, as well as a Pharmacovigilance Risk Assessment Committee (“PRAC”) Rapporteur and Co-Rapporteur. The Rapporteur and Co-Rapporteur are assigned with the primary responsibility of preparing and delivering an approvability evaluation report, supported by a team of assessors from their National Authority. In 2012, the PRAC Rapporteur and Co-Rapporteur was implemented, after the latest revisions to the EMA safety requirements on Pharmacovigilance and Risk Assessment Plan, with the primary responsibility of preparing and delivering an approvability evaluation report specifically with regards to safety. The EMA has also notified us regarding the EMA’s official designation of the Rapporteur and Co-Rapporteur, and PRAC Rapporteur and Co-Rapporteur to our planned MAA for BiovaxID. We conducted our EMA Pre-submission Meeting for the MAA on September 12, 2012, during which our planned application dossier was validated. Subsequently, on December 3, 2013, we submitted an MAA with the EMA for BiovaxID. We were formally notified the application had been validated by the EMA on January 7, 2014, confirming that the submission was complete, and beginning the formal EMA review process intended to secure approval to market BiovaxID in the EU and to allow prescription and sale of BiovaxID for the treatment of non-Hodgkin’s lymphoma in patients who have achieved a first complete remission.
Additionally, based on a scientific advice meeting conducted with Health Canada, we have announced plans to file a NDS seeking regulatory approval in Canada.
We conducted a formal guidance meeting with the FDA in order to discuss the path for our filing of a BLA for BiovaxID’s U.S. regulatory/marketing approval. As a result of this guidance meeting, we plan to conduct a second Phase 3 clinical trial to complete the clinical data gained through our first Phase 3 clinical trial and our BiovaxID development program to support our filing of our BLA for BiovaxID. We are preparing to initiate this second Phase 3 clinical trial subject to required funding.
As we continue to advance our efforts to comply with various regulatory validations and comparability requirements related to our manufacturing process and facility, no assurances can be given that substantial additional requirements will not be imposed by any regulatory agencies, including the EMA, Health Canada, and the FDA for the regulatory/marketing approval of BiovaxID.
Subsequent regulatory/marketing approval of BiovaxID, if any, may require us to perform additional clinical studies as a condition to approval or continued marketing of BiovaxID, which may result in additional clinical trial expenses. Once received, there can be no assurances that we will receive continued regulatory/marketing approval. Our ability to timely access required financing will continue to be essential to support the ongoing efforts to pursue the development and potential regulatory/marketing approval and commercialization efforts of BiovaxID.
Proprietary Rights to BiovaxID
As a result of the FDA’s Orphan Drug designation of BiovaxID for the treatment of FL, MCL and Waldenstrom's Macroglobulinemia, a rare B-cell subtype of NHL, we have 7 years of market exclusivity in the U.S. from the date of the FDA’s marketing approval for these three B-cell subtypes of NHL. We also have 10 years of market exclusivity in the EU as a result of EMA’s Orphan Medicinal Product designation of BiovaxID for the treatment of FL and MCL.
In addition, the regulations adopted in both the U.S. and the EU governing “biosimilar” products (the term adopted to describe generic biologic pharmaceutical products) provide us with “data exclusivity” (i.e., no biosimilar could reference the BiovaxID clinical data) for 12 years in the U.S. and 8 years in the EU. Those same biosimilar regulations make it extremely difficult to qualify as a “biosimilar”, and even for those products which can clear that hurdle independent clinical data is required prior to licensure.
In addition to market exclusivity based on governmental regulation, we rely on proprietary rights provided by a combination of an exclusive world-wide license to the cell line that is used in the production of BiovaxID, patent protection, trade secret protection, and our ongoing innovation. Although the composition of BiovaxID, in its current form, is not patentable, we have filed U.S. and foreign patent applications relating to methods of treatment using BiovaxID. We have also filed an international patent application (“PCT”) and a provisional application relating to methods for producing and selecting idiotype vaccines for treatment of B-cell cancers. Also, we have filed a provisional application covering the use of a biomarker for predicting cancer vaccine effectiveness and patient outcomes. In addition, we have filed U.S. and foreign patent applications relating to certain features of the AutovaxID® instrument used in the production of BiovaxID. The AutovaxID instrument is our proprietary production system which is fully enclosed, automated and has disposable components for each patient’s personalized vaccine. We believe that, without the availability of an automated production system, the methods used to produce a patient-specific immunotherapy are time-consuming and labor-intensive, resulting in a very expensive process that would be difficult to scale up. Following the findings related to the apparent role of the IgM isotype in clinical benefit from the vaccine, we filed a broad range of patent applications covering various applications of these findings. We have re-filed for U.S. registration of the trademark BiovaxID and have registered BiovaxID® as a Community Trademark in the EU. BiovaxID is manufactured with a proprietary cell line, which we have licensed on a worldwide exclusive basis from Stanford University (“Stanford”). We believe that the use of any cell line other than our exclusively licensed cell line, in the production of a similar idiotype vaccine would require filing a separate IND application and undergoing clinical testing evaluation by the FDA.
BiovaxID Manufacturing Process and Facility
Manufacturing Process
The BiovaxID manufacturing production process begins when a sample of the patient’s tumor is extracted by a biopsy and the sample is shipped refrigerated to our facility in Minneapolis (Coon Rapids), Minnesota. At our facility, we identify the idiotype that is expressed on the surface of the patient’s tumor cells through laboratory analysis. Additionally, we identify whether the isotype is IgM or IgG. In NHL, the tumor B-cells bear the surface idiotype (immunoglobulin or antibody) derived from the original transformed malignant B-cell, but do not typically secrete it in an amount suitable for vaccine production. In order to make sufficient quantities of idiotype for vaccination, the patient’s tumor cells are then fused with an exclusively licensed cell line (mouse/human heterohybridoma cell line, K6H6) from Stanford to create a hybridoma or hybrid cell.
After the creation of the hybridoma, we determine which hybridoma cells display the same antigen idiotype as the patient’s tumor cells, and those cells are selected to produce the vaccine. The selected hybridoma cells are then seeded into our proprietary hollow fiber bioreactors, where they are cultured and where they secrete or produce idiotype antigen. The secreted idiotype is then collected from the cells growing in the hollow fiber bioreactor. After a sufficient amount of idiotype is collected for the production of an appropriate amount of the vaccine, the patient’s idiotype is purified using multi-step purification processes (see Figure 10a).
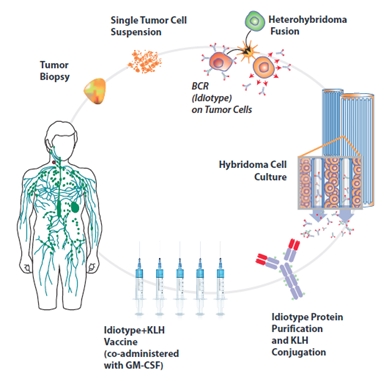
Figure 10a. Individualized Manufacturing Process for BiovaxID Immunotherapy: (Clockwise) Beginning with an excisional (>2cm) lymph node biopsy, tumor cells are fused with our proprietary mouse/human heterohybridoma in order to induce secretion of normally surface-bound tumor immunoglobulin (idiotype). Id-secreting clones are identified by comparing their unique idiotype sequence to the tumor’s after which they are cultured (expanded) in a proprietary hollow fiber bioreactor system (not shown). During culture, supernatant (containing idiotype) is collected until sufficient amounts have been produced to yield adequate dosage of vaccine. This supernatant is purified by affinity chromatography and conjugated (bonded) to KLH carrier protein, resulting in a finished vaccine that can be shipped and administered to patients. In our Phase 3 clinical trial, manufacturing success was approximately 95% of treated patients. (Fig. reprinted from Neelapu, et al. Exp. Opin Biol Ther 2007).
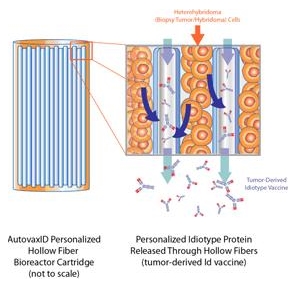
Figure 10b. Hollow fiber perfusion to produce the cell cultures used in the manufacture of BiovaxID.
We use a method known as “hollow fiber perfusion” to produce the cell cultures used in the manufacture of BiovaxID (see Figure 10b). Hollow fiber perfusion, as compared to other cell culture methods, seeks to grow cells to higher densities more closely approaching the density of cells naturally occurring in body tissue. The hollow fiber perfusion method involves using hair-like plastic fibers with hollow centers which are intended to simulate human capillaries. Thousands of these fibers are inserted in a cartridge, which we refer to as a bioreactor. The cells are grown on the outside of the hollow fibers while nutrient media used to support cell growth is delivered through the hollow centers of the fibers. The fiber walls have small pores, allowing nutrients to pass from the hollow center to the cells. The fibers act as filters and yield concentrated secreted products. Because the cells are immobilized in the bioreactor, the concentrated product can be harvested during the ongoing cell growth process. We believe that hollow fiber technology permits the harvest of cell culture products with generally higher purities than stirred-tank fermentation, a common alternative cell culture method, thereby reducing the cost of purification as compared to stirred tank fermentation. Additionally, the technology associated with the hollow fiber process generally minimizes the amount of costly nutrient media required for cell growth as opposed to other cell culturing techniques.
After manufacture and purification, the resulting purified idiotype is then conjugated, or joined together, with KLH, to create the vaccine. KLH is a foreign carrier protein that is used to improve the immunogenicity, or ability to evoke an immune response, of the tumor-specific idiotype. The BiovaxID vaccine is then frozen and shipped to the treating physician. At the treating physician’s office, the vaccine is thawed and injected into the patient.
BiovaxID is administered in conjunction with GM-CSF, a natural immune system growth factor that is administered with the idiotype vaccine to stimulate the immune system and increase the response to the idiotype vaccine. In the Phase 2 and Phase 3 clinical trials FL patients were administered 5 monthly BiovaxID injections in the amount of 0.5 milligram of idiotype per injection, with the injections being given over a 6-month period of time in which the fifth month is skipped. Through this process, the patient-specific idiotype is used to stimulate the patient’s immune system into targeting and destroying malignant B-cells bearing the same idiotype.
We estimate that an average of 2 to 3 months is required to manufacture each vaccine, which for most patients may overlap the time period when induction chemotherapy is being administered. While the manufacturing process for BiovaxID is highly personalized to each patient, we consider it to be highly controlled and predictable. The most common reason for a failure to successfully produce a patient’s vaccine was the presence of rare idiotype variants as opposed to the failure of a step in the manufacturing process. During the Phase 3 clinical trial, we experienced approximately 95% success rate in manufacturing our BiovaxID vaccines.
Manufacturing Facility
BiovaxID is a personalized medicine which is produced separately for each individual patient through a laboratory-type process based on the patient’s own tumor cells derived by biopsy. Following regulatory/marketing approval of BiovaxID, we plan to initially produce BiovaxID in our existing leasehold space in Minneapolis (Coon Rapids), Minnesota. In order to facilitate the regulatory/marketing approval process, we have completed a dedicated pilot scale suite of laboratory clean rooms especially designed to produce BiovaxID. As the regulatory/marketing approval process advances toward completion and subject to availability of funding, we anticipate expanding our current leasehold space or adding new manufacturing facilities as required to, meet the anticipated commercialization requirements. During our Phase 3 clinical trial, BiovaxID was produced at our facility in Worcester, Massachusetts. Because, we have relocated the site of the manufacturing process to our Minneapolis (Coon Rapids), Minnesota facility following the Phase 3 clinical trials and because we are expanding this facility, we are currently in the process of demonstrating to the national and/or international regulatory agencies that the BiovaxID vaccine produced under these new conditions is comparable to the BiovaxID vaccine that was the produced subject of earlier clinical testing. This requirement will also apply to future expansions of the facility, such as the possible expansion to additional facilities that may be required for successful commercialization of BiovaxID. There is also a requirement for validation of the manufacturing process for BiovaxID utilizing our AutovaxID® instrument. A showing of comparability requires data demonstrating that the BiovaxID vaccine produced continues to be safe, pure, and potent and may be based on chemical, physical, and biological assays and, in some cases, other non-clinical data.
INSTRUMENTS AND DISPOSABLES
We provide a range of cell culture systems that are used to produce small, research and development-scale through large, production-scale quantities of cell culture products. Our cell culture systems all incorporate perfusion technology and hollow fiber cartridges. This segment of our business represented approximately $1.6 million (approximately 39%) and $0.8 million (approximately 22%) of our tatal revenues for the years ended September 30, 2013 and 2012, respectively. Because of these shared characteristics, all of our cell culture systems listed below can be used to culture a variety of cell lines (hybridomas, CHO, MDCK, BHK and others) to produce a range of cell-based products, such as monoclonal antibodies, other secreted proteins, virus, virus-like particles, vaccines, and whole cells. Our smaller cell culture systems can be scaled-up to any of our larger cell culture systems. Our cell culture system product line, in order of increasing capacity, includes:
HF PRIMER™: The HF Primer is a low-cost cell culture system capable of producing small, research and development quantities of the cell culture products from mammalian cell lines. The HF Primer also provides a relatively inexpensive option to evaluate the efficacy of new cell lines or applications in perfusion technology that can then be scaled-up to any of our larger cell culture systems. HF Primer is a single-use, fully disposable product that requires no investment in custom equipment.
MULTI-6™: The Multi-6 is a low-cost cell culture system capable of simultaneously producing small, research and development -scale quantities of six different cell culture products. Optionally, the Multi-6 can be used to produce one cell culture product at a rate that is six-fold higher than the HF Primer. This flexibility allows a researcher to operate one Multi-6 instead of managing six separate HF Primer systems running side-by-side. Multi-6 applications can be scaled-up to our larger AutovaxID® or other cell culture systems listed below, if the need arises. Multi-6 is a single-use, fully disposable product that requires no investment in custom equipment.
AUTOVAXID®: The AutovaxID is our most advanced, fully automated cell culture system. Its setup and operation requires very little technician expertise and labor in comparison to competing cell culture system technologies and even our other cell culture systems listed below. The AutovaxID was designed from the beginning to: enable multi-product facilities, capture most all production information into its CFR 21 Part 11-compliant electronic records system, and operate as a fully closed-system biologics-manufacturing platform. The AutovaxID allows supervisors to implement built-in software controls to ensure technicians adhere to strictly defined production protocols for cGMP environments or remove this feature when it is not needed, such as in research and development operations. There are four single-use AutovaxID disposables to provide researchers the needed flexibility to go from research and development- through pilot-scale production. One of the disposable options even allows the simultaneous culturing of three different cell lines to produce three different products in small quantities. When needed, one cell line can then be scaled-up to AutovaxID’s larger-capacity disposables. We plan to utilize AutovaxID’s advanced capabilities to streamline commercial manufacture of BiovaxID. AutovaxID is the first cell culture system that enables production of personalized cell-based treatments economically and in compliance with cGMP standards. Additionally, we have contracted with the DoD and others to further develop the AutovaxID and related hollow fiber bioreactor systems to explore potential production of additional vaccines, including vaccines for viral indications such as influenza and other contagious diseases.
ACUSYST-MINIMAX®: The miniMAX is an economical system that provides the flexibility and technology needed to support optimization studies and research- through pilot-scale production of the cell culture products listed above. The miniMax is a tabletop instrument that uses simple, but robust, microprocessors to automatically control pH, incubator temperature, fluid-flow dynamics, and seven process pumps. This system offers two single-use disposables options to meet varying production requirements. Optimization results determined in the miniMAX can be transferred to our larger systems listed below. The production capacity is equivalent to an AutovaxID, but the miniMAX does not have the advanced features, controls, simpler setup and ease of operation available through the AutovaxID.
ACUSYST-MAXIMIZER®: The Maximizer is an economical system with a design that is similar to the miniMAX system. This system has all of the features of miniMAX, using single-use disposables that either match or double the production capacity of the miniMAX system. The Maximizer is an economical system for process development and routine productions. The Maximizer offers the flexibility of four single-use disposables to meet varying production requirements. Production processes determined in the Maximizer can be scaled-up to our largest cell culture system, AcuSyst-Xcellerator®.
ACUSYST-XCELLERATOR®: The Xcellerator is a freestanding, production-scale cell culture system that has been used for production of a FDA-licensed biologic. The Xcellerator contains an incubator, a refrigerator, a built-in computer that has a CFR 21 Part 11-compliant electronic records system, numerous process pumps, and a touch screen for local control. The Xcellerator supports remote monitoring and/or control via standard web browsers. The system supports two independent single-use disposables and, therefore, can simultaneously culture two different cell lines to produce two products. The Xcellerator has a production capacity that is equivalent to a 20x scale-up of AutovaxID’s capacity.
In addition to instrument and disposables sales, we have recurring revenue from the sale of hollow fiber bioreactors, cultureware, tubing sets and other disposable products and supplies for use with our cell culture system instrument product line.
Currently, we assemble, validate and package the instruments and disposables, cell culture products and services which we sell. Customers for our instruments and disposables are the same potential customers targeted for our contract cell culture products and services which include biopharmaceutical and biotechnology companies, medical schools, universities, research facilities, hospitals and public and private laboratories.
Proprietary Rights to Instruments and Disposables
We own several patents covering various aspects of our hollow fiber perfusion process, instruments and proprietary cell culturing methods. We own U.S. Patent No. 6,001,585, which protects the HF Primer™ cell culture system and its use in screening cell lines and process conditions. Although the HF Primer is impractical for large-scale vaccine production, it may be used as an efficient screening tool to cost-effectively determine how well a cell line will perform in a hollow fiber system, such as the AutovaxID® instrument, which is used in the production of BiovaxID. U.S. Patent No. 6,001,585 will expire in November 2017.
We have also filed U.S. and foreign patent applications relating to certain features of the AutovaxID instrument. Several patents have been granted in the EU covering the AutovaxID’s extra-capillary (“EC”) fluid cycling system, which enables control of fluid volumes in the hollow fiber bioreactor during manufacture of BiovaxID in a closed, contamination-free environment. These EU patents will remain in force until 2027.
We have filed (a) a PCT application covering the use of AutovaxID instrumentation for rapid, large-scale production of virus, virus-like particles, and viral vaccines at a high yield, (b) a PCT application concerning an integrated apparatus and method for production and purification of antibodies, and (c) a provisional application concerning cultureware modules and biomanufacturing suites for large-scale production of cells and cell-derived products such as antibodies, proteins, virus, and virus-like particles.
CELL CULTURE PRODUCTS AND SERVICES
We manufacture mammalian cell culture products such as, whole cells, recombinant and secreted proteins, and monoclonal antibodies. Additionally, we provide related services as a contract resource to assist our customers in developing cell production process protocols, cell line optimization, cell culture production optimization, media evaluation and other related services. This segment of our business represented approximately $1.6 million (approximately 39%) and $0.8 million (approximately 22%) of our total revenues for the years ended September 30, 2013 and 2012, respectively.
With our focus on pre-launch therapeutics and marketed diagnostics production, we serve all the manufacturing needs of our customers. From process development to regulatory support, our one-stop comprehensive approach provides solutions for clients that have chosen to temporarily or permanently outsource any or all stages of drug development. If needed, commercial production capacity for product launch is seamless, we provide all the required regulatory support, including biocompatibility studies.
With nearly 30 years of expert experience in cell culture, we formulate the optimal strategy for our customers’ biologics manufacturing. By accurately matching the level of hollow fiber bioreactor production technology to our customers’ phase of product development, our customers can manage their overall investment risk. Only as products advance to the next stage of development do we scale to higher-output production systems. Customers of our cell culture products and services are biopharmaceutical and biotechnology companies, medical schools, universities, research facilities, hospitals and public and private laboratories. We generally produce cell culture products pursuant to contracts which specify the customer’s requirements for the cell culture products to be produced or the services to be performed.
There are various processes commonly used to expand mammalian cells generally used for the production of antibodies. These may include hollow fiber bioreactor perfusion, stirred tank fermentation, roller bottle and other processes. We primarily use hollow fiber bioreactor technology to expand customer provided cell lines and produce the respective monoclonal antibodies. This technology grows cells to higher densities which more closely mimics mammalian physiology. We have significant expertise with in vitro (outside the living body) cell culture methods for a wide variety of mammalian cells. Mammalian cells are complicated and dynamic, with constantly changing needs. A primary component of hollow fiber bioreactors is fibers made of plastic polymers. The fibers are hair-like with hollow centers which simulate human capillaries. Thousands of these fibers are inserted in a cartridge, which we refer to as a bioreactor. The cells are grown on the outside of the hollow fibers while nutrient media used to support cell growth is perfused through the lumen of the fibers. The fiber walls have small pores, allowing nutrients to pass from the hollow center to the cells. The fibers act as filters and yield concentrated secreted products. Because the cells are immobilized in the bioreactor, the concentrated product can be harvested during the ongoing cell growth process. Hollow fiber technology permits harvests of cell culture products with generally higher purities thereby reducing the cost of downstream purification processes. This technology generally minimizes the amount of costly nutrient media required for cell growth.
The most generally used process for mammalian cell production is stirred tank fermentation. Hollow fiber bioreactor technology can be contrasted with the competitive stirred tank fermentation process which takes place in tanks of various sizes. Cells are grown inside the tanks in culture medium which is maintained under controlled conditions and continuously stirred to stimulate growth. At the end of the growing process, as opposed to incrementally during the growth process, cells are separated from the medium and the protein of interest is isolated through a series of complex purification processes. The size of the tanks generally result in stirred tank fermentation facilities requiring significantly more start-up costs, space and infrastructure than comparable production facilities using hollow fiber technology. While stirred tank fermentation and hollow fiber technology are both used for cell production of various quantities, we believe that the stirred tank fermentation process is currently more commonly used for larger scale commercial production requirements. We believe that hollow fiber technology has advantages in scalability, start-up time and cost in the early development of antibody production.
Our patented hollow fiber technology is the key to optimizing our customers’ biologics manufacturing. For Phase I, Phase II and Phase III cGMP production, our hollow fiber bioreactor perfusion technology provides economic advantages through largely automated culture of a relatively large, densely packed population of cells in a small space. Additionally, the low molecular weight cut-off of the fiber membrane allows more expensive media components to be conserved, while less expensive basal media is continuously perfused through the bioreactor. As a result, this automated approach can be more cost-effective than conventional platforms generally used for maintenance of large cell populations. In the expanding field of personalized medicine where patient specific drugs and therapeutics are frequently envisioned, such as BiovaxID, we believe that hollow fiber technology may be the appropriate cell culture production technology.
COMPETITION FOR BIOVAXID
Biotechnology has experienced, and is expected to continue to experience, rapid and significant change. The use of monoclonal antibodies as initial or induction therapy, and increasingly for maintenance therapy, has become well-established and generally accepted. Products that are well-established or accepted, including monoclonal antibodies such as Rituxan®, may constitute significant barriers to market penetration and regulatory approval which may be expensive, difficult or even impossible to overcome. New developments in biotechnological processes are expected to continue at a rapid pace in both industry and academia, and these developments are likely to result in commercial applications competitive with BiovaxID. We expect to encounter intense competition from a number of companies that offer products in our targeted application area. We anticipate that our competitors in these areas will consist of both well-established and development-stage companies and will include:
|
|
•
|
healthcare companies;
|
|
|
•
|
chemical and biotechnology companies;
|
|
|
•
|
biopharmaceutical companies; and
|
|
|
•
|
companies developing drug discovery technologies.
|
We expect to compete on, among other things, the safety and efficacy of our product candidates and more desirable treatment regimens, combined with the effectiveness of our experienced management team. Competing successfully will depend on our continued ability to attract and retain skilled and experienced personnel, to identify and secure the rights to and develop pharmaceutical products and compounds and to exploit these pharmaceutical products and compounds commercially before others are able to develop competitive products.
If approved, BiovaxID will be required to compete with currently approved therapies, as well as therapies which may be approved in the future. There are currently no approved active immunotherapeutic drugs which seek to induce an adaptive, specific and durable immune response to identify and eradicate the residual lymphoma cells remaining after a patient achieves remission in an effort to extend that remission or avoid relapse. BiovaxID is a therapy designed to be administered to lymphoma patients who have achieved complete remission after initial chemotherapy treatment. If approved, BiovaxID would represent a new class of drugs available to treat FL and potentially offering a new treatment option for FL patients.
BiovaxID is the only personalized cancer vaccine for treatment of FL that has demonstrated significant clinical benefit in a Phase 3 clinical trial. Two other vaccines, MyVaxTM developed by Genitope and Specifid™ developed by Favrille which were studied in Phase 3 clinical trials in FL patients did not report statistically significant clinical benefit and we believe are no longer under development. There are fundamental structural differences between BiovaxID and the cancer vaccines developed by Genitope and Favrille; Genitope and Favrille manufactured their respective vaccines with IgG isotypes without regard to the patient’s actual isotype and their clinical trial designs under which the clinical efficacy of these vaccines were tested were different, which we believe explain why BiovaxID achieved significant clinical benefit while Genitope and Favrille’s vaccines did not.
Chemotherapy and monoclonal antibodies are widely used for the treatment of FL. Although chemotherapy and monoclonal antibodies can substantially reduce the tumor mass and in most instances achieve clinical remission, the remission is generally of limited duration. FL patients generally relapse and the cancer usually becomes increasingly resistant to further chemotherapy treatments. The patient’s response to therapy becomes briefer and weaker with each additional course of therapy, such that eventually further chemotherapy would offer no clinical benefit.
A number of passive immunotherapies, such as rituximab and radioimmunotherapeutic agents (radioisotopes linked to monoclonal antibodies), are approved by the FDA for the treatment of FL. A monoclonal antibody is a type of antibody produced in large quantity that is specific to an antigen that is expressed by tumor cells and also by some normal cells. These therapies have been used as primary treatment and also as part of combination induction therapy including chemotherapy and rituximab based therapy is considered to be the standard of care to treat FL. In an effort to prolong the duration of the clinical remission monoclonal antibodies have increasingly been used as maintenance therapies.
If approved to treat FL, BiovaxID will face competition from other approved drugs, including rituximab maintenance. Penetrating a market and achieving usage by physicians and patients in the face of an established standard of care is anticipated to represent a significant marketing challenge.
If approved to treat MCL, BiovaxID will be required to compete with other approved and/or development therapies for the treatment of MCL. There is currently no consensus standard of care for the first line treatment of MCL; however, there are a number of FDA-approved agents used for the treatment of MCL both in first line settings and in patients in relapse.
COMPETITION FOR AUTOVAXID®
There are many kinds of technologies for the manufacture of cell-based products. The technology relied upon by our instruments and disposables referred to as hollow fiber perfusion which is not widely accepted for large-scale manufacturing and, notwithstanding our development efforts, may not become widely accepted in the future. Our hollow fiber bioreactors must compete with many other kinds of cell-based manufacturing instruments including, but not limited to, stirred-tank reactors; airlift fermentors; roller bottles; packed bed reactors; two-chamber reactors; ceramic matrix systems; batch fermentation techniques; and WAVE bioreactors. There can be no assurance that our hollow fiber systems will gain widespread acceptance competing with other established technologies marketed by industry leaders such as General Electric, Lonza, 3M, Sartorius Stedim, Thermo Scientific and Sandoz Biopharmaceuticals.
Competition for cell-based instruments is intense. Most of the developers, manufacturers and marketers of cell-based manufacturing instruments are much larger, more entrenched with potential customers and better financed than us which places us at a competitive disadvantage.
PATENTS, TRADEMARKS AND PROTECTION OF PROPRIETARY TECHNOLOGY
We are pursuing a number of methods to establish and maintain market exclusivity for our product candidates to the greatest extent possible, including seeking patent protection, the use of statutory market exclusivity provisions, and otherwise protecting our intellectual property.
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology, and know-how; to operate without infringing the proprietary rights of others; and to prevent others from infringing on our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications when possible relating to our proprietary technology, inventions, and improvements that are important to our business. We also rely on trade secrets, know-how, continuing technological innovation, and in-licensing opportunities to develop and maintain our proprietary position. The following is information regarding our owned and licensed patents and patent applications that we consider material to our business:
We own several patents covering various aspects of our hollow fiber perfusion process, instruments and proprietary cell culturing methods. Our patents also cover aspects of our therapeutic vaccine production process. We plan to continue pursuing patent and other intellectual property protection for our product candidates, our technology and know-how. Currently, we have three issued U.S. patents. Additionally, we have several patent applications that are pending. Our presently issued U.S. patents will expire in November 2017 and July 2029. A list of our U.S. and foreign patents and published patent applications are as follows:
|
Patent No.
|
Title and Inventor(s)
|
Filing Date/Issue Date
|
Expiration Date
|
|||
|
6,001,585
|
MICRO HOLLOW FIBER BIOREACTOR by Michael J. Gramer
|
Nov. 14, 1997/Dec 14, 1999
|
Nov. 14, 2017
|
|||
|
8,383,397
|
Method and System for the Production of Cells and Cell Products and Applications Thereof by Robert J. Wojciechowski et al.
|
Nov. 20, 2008/February 26, 2013
|
July 30, 2029
|
|||
|
8,540,499
|
Extra-Capillary Fluid Cycling System and Method for a Cell Culture Device by Darrell P. Page et al.
|
Nov. 20, 2008/Sept. 24, 2013
|
July 4, 2029
|
|
Foreign Patent No.
|
Title and Inventor(s)
|
Filing Date/Issue Date
|
Expiration
Date
|
|||
|
EP 2027247 (UK)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
Nov. 20, 2008/Jan. 26, 2011
|
May 21, 2027
|
|||
|
DE602007012238D (Germany)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
Nov. 20, 2008/Jan. 26, 2011
|
May 21, 2027
|
|||
|
AT2027247 (Austria)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
Nov. 20, 2008/Jan. 26, 2011
|
May 21, 2027
|
|||
|
P2027247 (Switzerland)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
Nov. 20, 2008/Jan. 26, 2011
|
May 21, 2027
|
|||
|
FR2027247 (France)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
Nov. 20, 2008/Jan. 26, 2011
|
May 21, 2027
|
|||
|
GB2027247 (Great Britain)
|
EXTRA-CAPILLARY FLUID CYCLING SYSTEM AND METHOD FOR A CELL CULTURE DEVICE by Darrell P. Page et al.
|
May 21, 2007/Jan. 26, 2011
|
May 21, 2027
|
|||
|
HK1166343 (Hong Kong)
|
METHOD AND SYSTEM FOR THE PRODUCTION OF CELLS AND CELL PRODUCTS AND APPLICATIONS THEREOF by Robert J. Wojciechowski et al.
|
May 21, 2007/October 26, 2012
|
|
Application
Publication No.
|
Title and Inventor(s)
|
Filing Date/Publication Date
|
Countries/Regions
|
|||
|
WO 2012/021840
|
MATERIALS AND METHODS FOR DESIGNING AUTOLOGOUS IDIOTYPE VACCINES AND TREATMENT OF B-CELL MALIGNANCIES by Carlos Santos et al.
|
Aug. 12, 2011/Feb. 16, 2012
|
International
|
|||
|
WO 2012/064760
|
MATERIALS AND METHODS FOR DIRECTING AN IMMUNE RESPONSE AGAINST AN EPITOPE by Carlos Santos et al.
|
Nov. 9, 2011/May 18, 2012
|
International
|
|||
|
EP 2637691
|
MATERIALS AND METHODS FOR DIRECTING AN IMMUNE RESPONSE TO AN EPITOPE by Carlos Santos et al.
|
Nov. 8, 2011/Sept. 18, 2013
|
Europe
|
|||
|
WO 2012/171026
|
METHOD AND APPARATUS FOR VIRUS AND VACCINE PRODUCTION by Mark Hirschel et al.
|
June 11, 2012/Dec. 13, 2012
|
International
|
|||
|
WO 2013/086418
|
TUMOR-SPECIFIC GM-CSF CYTOKINE RESPONSE AS PREDICTOR OF CANCER VACCINE EFFECTIVENESS by Wyndham H. Wilson et al.
|
Dec. 7, 2012/June 13, 2013
|
International
|
|||
|
US 2013/0058907
|
METHOD AND SYSTEM FOR THE PRODUCTION OF CELLS AND CELL PRODUCTS AND APPLICATIONS THEREOF by Robert J. Wojciechowski et al.
|
October 30, 2012/March 7, 2013
|
United States
|
|||
|
WO 2012/171030
|
METHOD AND APPARATUS FOR ANTIBODY PRODUCTION AND PURIFICATION by Mark Hirschel et al.
|
June 11, 2012/Dec. 13, 2012
|
International
|
We have filed PCT applications based on or related to various aspects of our analyses of clinical benefit based on isotype, and to sequencing results matching vaccine to antigen coverage for the development of a companion diagnostic and to use of the AutovaxID® instrument for the production of viruses, virus-like particles, and antiviral vaccines such as those targeting influenza and other contagious diseases. In addition to its independent research and development programs, it is anticipated that our collaborations with industry and research partners will generate additional valuable intellectual property, which will be wholly-owned, jointly owned and/or licensed to us.
We also possess licensed intellectual property used in the development and manufacture of BiovaxID. BiovaxID is manufactured with a proprietary cell line, which we have licensed on a world-wide exclusive basis from Stanford. This is significant, because we believe that the use of any cell line other than our exclusively licensed cell line, in the production of a similar idiotype vaccine, would require filing a separate IND application and undergoing clinical testing evaluation by the FDA.
Additionally, we consider trademarks to be important to our business. We have established trademarks covering various aspects of our hollow fiber perfusion process, instruments and proprietary cell culturing methods (AutovaxID®, Acusyst-Maximizer® and Acusyst-Xcell®). We have applied for the U.S. and foreign registration of the trademark BiovaxID in connection with our therapeutic cancer vaccine and Autovax™ in connection with our instrument used in the manufacture of BiovaxID. We plan to continue aggressively pursuing trademark and other proprietary protection for our therapeutic vaccine technology and instrumentation, both nationally and internationally.
Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated, or circumvented, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our product candidates. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies or duplicate any technology developed by us. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
We rely in some circumstances on trade secrets to protect our technology, particularly with respect to certain aspects of our BiovaxID manufacturing process. However, trade secrets are difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors, and other contractors. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
GOVERNMENT REGULATION
Government authorities in the U.S. at the federal and state local levels and in foreign countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, sampling, marketing, and import and export of pharmaceutical products, biologics, and medical devices. All of our product candidates in development will require regulatory and/or marketing approval by government agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical trials and other approval procedures of the FDA and similar regulatory authorities in foreign countries. Various federal, state, local, and foreign statutes and regulations also govern testing, manufacturing, safety, labeling, storage, and record-keeping related to such products and their marketing. The process of obtaining these approvals and the subsequent process of maintaining substantial compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. The FDA regulates drugs and well-characterized biologics under the Federal Food, Drug, and Cosmetic Act (“FDCA”), and implementing regulations that are adopted under the FDCA. In the case of biologics, the FDA regulates such products under the Public Health Service Act. If we fail to comply with the applicable requirements under these laws and regulations at any time during the product development process, approval process, or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawals of approvals, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of its operations, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us. The FDA also administers certain controls over the export of drugs and biologics from the U.S. Any failure by us, our suppliers of manufactured drug product, collaborators or licensees to obtain or maintain, or any delay in obtaining, regulatory approvals could adversely affect the marketing of our product candidates and our ability to receive product revenue, license revenue or profit sharing payments. In addition, statutes, rules, regulations, and policies may change and new legislation or regulations may be issued that could delay such approvals.
The FDA has extensive regulatory authority over biopharmaceutical products (drugs and diagnostic products produced from biologic processes). The principal FDA regulations that pertain to our cell production activity include, but are not limited to 21CFR Parts 600 and 610 – General Biological Products and Standards; 21 CFR Parts 210 and 211 – current Good Manufacturing Practices for Finished Pharmaceuticals; 21 CFR Part 820 – Quality System Regulations (medical devices); and 21 CFR Part 58 – Good Laboratory Practice for Non-Clinical Laboratory Studies. FDA’s guidelines include controls over procedures and systems related to the production of mammalian proteins and quality control testing of any new biological drug or product intended for use in humans (including, to a somewhat lesser degree, in vivo biodiagnostic products). FDA guidelines are intended to assure that the biological drug or product meets the requirements through rigorous testing with respect to safety, efficacy, and meet the purity characteristics for identity and strength. FDA approvals for the use of new biological drugs or products (that can never be assured) require several rounds of extensive preclinical testing and clinical investigations conducted by the sponsoring pharmaceutical company prior to sale and use of the product. At each stage, the approvals granted by the FDA include the manufacturing process utilized to produce the product. Accordingly, our cell culture systems used for the production of therapeutic or biotherapeutic products (biological drug or product) are subject to significant regulation by the FDA under the FDCA.
Our cell culture systems used to produce cells for diagnostic uses are regulated under the FDCA as Class I medical devices. Medical devices are classified by the FDA into three classes (Class I, Class II and Class III) based upon the potential risk to the consumer posed by the medical device (Class I medical devices pose the least amount of risk, while Class III medical devices and “new” devices are presumed to inherently pose the greatest amount of risk). As Class I medical devices, our systems must be manufactured in accordance with cGMP guidelines. Sales of such systems to customers using them to manufacture materials for clinical studies and licensure do not require prior FDA approval.
The process of complying with FDA guidelines and obtaining approvals from the FDA of applications to market biopharmaceutical drugs and products is costly, time consuming and subject to unanticipated delays. There is no assurance that our customers will be able to obtain FDA approval for biological drugs and products produced with our systems, and failure to receive such approvals may adversely affect the demand for our services.
Under the FDCA, our customers must establish and validate standard operating procedures (“SOPs”) utilizing our cell culture technologies in their drug master files. We provide assistance in operational, validation, calibration and preventive maintenance SOPs to customers, as needed, to support their product development and commercialization processes. For example, we will typically provide existing and prospective customers who are utilizing our contract production services or constructing production facilities based on our cell culture technologies with information to enable such customers to comply with the FDA’s guidelines required for facility layout and design. This information may be provided either in a drug/biologic master file that we give permission to customers to cross reference in their submission to the FDA, or provided to customers to include in their FDA submissions.
As we currently do business in a significant number of countries, in addition to the requirements of the FDA, we are subject to the regulations of other countries and their governmental agencies which apply to our goods and services when sold in their jurisdiction.
We are subject to various regulations regarding handling and disposal of potentially hazardous materials, wastes and chemicals such as cells and their secreted waste products, including those enforced by the U.S. Environmental Protection Agency (“EPA”) and various state and local agencies.
Pharmaceutical Product Regulation
Regulation by governmental authorities in the U.S. and other countries is a significant factor in the manufacture and marketing of pharmaceuticals and in our ongoing research and development activities. Most, if not all, of our product candidates require regulatory approval by governmental agencies prior to commercialization. In the case of biologics, they are subject to rigorous preclinical testing and clinical trials and other pre-marketing approval requirements by the FDA and foreign regulatory authorities.
Each of these three phases is discussed further below.
Preclinical Phase
The activities required before a product may be marketed in the U.S. and other countries begin with preclinical testing not involving human subjects. The development of a new pharmaceutical agent begins with the discovery or synthesis of a new molecule or well-characterized biologic. These agents are screened for pharmacological activity using various animal and tissue models, with the goal of selecting a lead agent for further development. Additional studies are conducted to confirm pharmacological activity, to generate safety data, and to evaluate prototype dosage forms for appropriate release and activity characteristics. Once the pharmaceutically active molecule is fully characterized, an initial purity profile of the agent is established. During this and subsequent stages of development, the agent is analyzed to confirm the integrity and quality of material produced. In addition, development and optimization of the initial dosage forms to be used in clinical trials are completed, together with analytical models to determine product stability and degradation. A bulk supply of the active ingredient to support the necessary dosing in initial clinical trials must be secured. Upon successful completion of preclinical safety and efficacy studies in animals, an IND submission is prepared and provided to the FDA for review prior to commencement of human clinical trials. The IND consists of the initial chemistry, analytical, formulation, and animal testing data generated during the preclinical phase. In general, the review period for an IND submission is 30 days, after which, if no comments are made by the FDA, the product candidate can be studied in Phase 1 clinical trials.
Clinical Phase
Following successful submission of an IND, the sponsor is permitted to conduct clinical trials involving the administration of the investigational product candidate to human subjects under the supervision of qualified investigators in accordance with good clinical practice. Typically, clinical evaluation involves the following time-consuming and costly three-phase sequential process:
|
|
•
|
Phase 1. Phase 1 clinical trials are conducted in a limited number of healthy individuals to determine the drug or biologics’ safety and tolerability and include biological analyses to determine the availability and metabolization of the active ingredient following administration. The total number of subjects and patients included in Phase 1 clinical trials varies, but are generally in the range of 20 to 80 people.
|
|
|
•
|
Phase 2. Phase 2 clinical trials involve administering the drug to individuals who suffer from the target disease or condition to determine the drug or biologics’ potential efficacy and ideal dose. These clinical trials are typically well controlled, closely monitored, and conducted in a relatively small number of patients, usually involving no more than several hundred subjects. These clinical trials require scale up for manufacture of increasingly larger batches of bulk chemical. These batches require validation analysis to confirm the consistent composition of the drug or biologics.
|
|
|
•
|
Phase 3. Phase 3 clinical trials are performed after preliminary evidence suggesting effectiveness of a drug or biologics has been obtained and safety (toxicity), tolerability, and an ideal dosing regimen have been established. Phase 3 clinical trials are intended to gather additional information about the effectiveness and safety that is needed to evaluate the overall benefit-risk relationship of the drug or biologics and to complete the information needed to provide adequate instructions for the use of the drug or biologics, also referred to as the Official Product Information. Phase 3 clinical trials usually include from several hundred to several thousand subjects.
|
Throughout the clinical phase, samples of the product made in different batches are tested for stability to establish shelf life constraints. In addition, large-scale production protocols and written standard operating procedures for each aspect of commercial manufacture and testing must be developed. Phase 1, 2, and 3 testing may not be completed successfully within any specified time period, if at all. The FDA closely monitors the progress of each of the three phases of clinical trials that are conducted under an IND and may, at its discretion, reevaluate, alter, suspend, or terminate the testing based upon the data accumulated to that point and the FDA’s assessment of the risk/benefit ratio to the patient. The FDA may suspend or terminate clinical trials at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. The FDA can also request additional clinical trials be conducted as a condition to product approval. Additionally, new government requirements may be established that could delay or prevent regulatory and marketing approval of our product candidates under development. Furthermore, institutional review boards, which are independent entities constituted to protect human subjects in the institutions in which clinical trials are being conducted, have the authority to suspend clinical trials at any time for a variety of reasons, including safety issues.
New Drug Application or Biologics License Application
After the successful completion of Phase 3 clinical trials, the sponsor of the new drug submits a NDA or BLA, in the case of biologics, to the FDA requesting approval to market the product for one or more indications. A NDA, or BLA, is a comprehensive, multi-volume application that includes, among other things, the results of all preclinical and clinical studies, information about the drug’s composition, and the sponsor’s plans for producing, packaging, and labeling the drug. Under the Pediatric Research Equity Act of 2003, an application also is required to include an assessment, generally based on clinical study data, on the safety and efficacy of drugs for all relevant pediatric populations before the NDA is submitted. The statute provides for waivers or deferrals in certain situations. In most cases, the NDA or BLA must be accompanied by a substantial user fee. In return, the FDA assigns a goal of 10 months from acceptance of the NDA or BLA to return of a first “complete response,” in which the FDA may approve the product or request additional information.
The submission of the application is no guarantee that the FDA will find it complete and accept it for filing. The FDA reviews all NDAs and BLAs submitted before it accepts them for filing. It may refuse to file the application and request additional information rather than accept the application for filing, in which case, the application must be resubmitted with the supplemental information. After application is deemed filed by the FDA, the FDA reviews an NDA or BLA to determine, among other things, whether a product is safe and effective for its intended use. The FDA has substantial discretion in the approval process and may disagree with an applicant’s interpretation of the data submitted in its NDA or BLA. Drugs that successfully complete NDA or BLA review may be marketed in the U.S., subject to all conditions imposed by the FDA. Prior to granting approval, the FDA generally conducts an inspection of the facilities, including outsourced facilities, which will be involved in the manufacture, production, packaging, testing and control of the drug product for cGMP compliance. The FDA will not approve the application unless cGMP compliance is satisfactory. If the FDA determines that the marketing application, manufacturing process, or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and will often request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the marketing application does not satisfy the regulatory criteria for approval and refuse to approve the application by issuing a “not approvable” letter or a “complete response” letter requiring additional steps to be completed before approval.
Biologics (such as BiovaxID) differ from other drugs for human use in that they must include more detailed chemistry and manufacturing information. This is necessary to help ensure the purity and quality of the product, for example to help ensure that it is not contaminated by an undesired microorganism or by another contaminant. The sponsoring company’s manufacturing facility must also supply product specific facility information that outlines the method of manufacture of the biologic in significant detail, since slight variations can result in a different final product. Further, an inspection of the manufacturing facility is completed to assess the production process and facility since these aspects also have a significant impact on the safety and efficacy of the product.
Post-Approval Phase
If the FDA approves the NDA or BLA, as applicable, the pharmaceutical product becomes available for physicians to prescribe in the U.S. After approval, the product is still subject to continuing regulation by FDA, including record keeping requirements, submitting periodic reports to the FDA, reporting of any adverse experiences with the product, and complying with drug sampling and distribution requirements. In addition, the sponsor is required to maintain and provide updated safety and efficacy information to the FDA. The sponsor is also required to comply with requirements concerning advertising and promotional labeling. In that regard, advertising and promotional materials must be truthful and not misleading. The sponsor is also prohibited from promoting any non-FDA approved or “off-label” indications of products. Failure to comply with those requirements could result in significant enforcement action by the FDA, including warning letters, orders to pull the promotional materials, and substantial fines. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval.
Drug and biologics manufacturers and their subcontractors are required to register their facilities and products manufactured annually with FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA to assess compliance with cGMP regulations. Facilities may also be subject to inspections by other federal, foreign, state, or local agencies. In addition, approved biological drug products may be subject to lot-by-lot release testing by the FDA before these products can be commercially distributed. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance.
In addition, following FDA approval of a product, discovery of problems with a product or the failure to comply with requirements may result in restrictions on a product, manufacturer, or holder of an approved marketing application, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or effectiveness data may require changes to a product’s approved labeling, including the addition of new warnings and contraindications. Also, the FDA may require post-market testing and surveillance to monitor the product’s safety or efficacy, including additional clinical studies, known as Phase 4 clinical trials, to evaluate long-term effects.
Orphan Drug Designation and Exclusivity
Pursuant to the U.S. Orphan Drug Act, a sponsor may request that the FDA designate a drug intended to treat a "rare disease or condition" as an "orphan drug." The term "orphan drug" can refer to either a drug or biologic. A rare disease or condition is defined as one which affects less than 200,000 people in the U.S., or which affects more than 200,000 people, but for which the cost of developing and making available the product is not expected to be recovered from sales of the product in the U.S. In the U.S., Orphan Drug designation must be requested before submitting a NDA or BLA. Upon the approval of the first NDA or BLA for a drug designated as an orphan drug for a specified indication, the sponsor of that NDA or BLA is entitled to seven years of exclusive marketing rights in the U.S. for the orphan drug for the same indication unless the sponsor cannot assure the availability of sufficient quantities of the drug to meet the needs of persons with the disease. However, orphan drug status is particular to the approved indication and does not prevent another company from seeking approval of an off-patent drug that has other labeled indications that are not under orphan or other exclusivities. The period of orphan exclusivity is concurrent with any patent or other exclusivity that relates to the drug or biologic. Orphan drugs may also be eligible for federal income tax credits for costs associated with the drugs' development. In order to increase the development and marketing of drugs for rare disorders, regulatory agencies outside the U.S., such as the EU (which has a ten year exclusivity marketing period) have enacted regulations similar to the Orphan Drug Act.
Orphan Drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If a product which has an Orphan Drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the product is entitled to a marketing exclusivity. For seven years, the FDA may not approve any other application, including NDAs or BLAs, to market the “same drug” for the same indication. The only exceptions are (i) where the second product is shown to be “clinically superior” to the product with Orphan Drug exclusivity, as that phrase is defined by the FDA and (ii) if there is an inadequate supply.
Manufacturing
Among the conditions for regulatory/marketing approval the regulatory agency, such as the FDA, the EMA and Health Canada, requires that the prospective manufacturer's quality control and manufacturing procedures continually conform to cGMP regulations (which are regulations governing the manufacture, processing, packing, storage and testing of drugs and biologics intended for human use). In complying with cGMP, manufacturers must devote extensive time, money and effort in the area of production and quality control and quality assurance to maintain full technical compliance. Manufacturing facilities and company records are subject to periodic inspections by the regulatory agency to ensure compliance. If a manufacturing facility is not in substantial compliance with these requirements, regulatory enforcement action may be taken by the regulatory agency, which may include seeking an injunction against shipment of products from the facility and recall of products previously shipped from the facility. Changes to the manufacturing process or site during or following the completion of clinical trials requires sponsors to demonstrate to the regulatory agency that the product under new conditions is comparable to the product that was the subject of earlier clinical testing. This requirement applies to relocations or expansions of manufacturing facilities, or additional facilities that may be required upon successful commercialization of the product. A showing of comparability requires data demonstrating that the product continues to be safe, pure, and potent and may be based on chemical, physical, and biological assays and, in some cases, other non-clinical data.
Medical Device Regulation
New medical devices are also subject to FDA approval and extensive regulation under the FDCA. Under the FDCA, medical devices are classified into one of three classes: Class I, Class II, or Class III. The classification of a device into one of these three classes generally depends on the degree of risk associated with the medical device and the extent of control needed to ensure safety and effectiveness.
Class I devices are those for which safety and effectiveness can be assured by adherence to a set of general controls. These general controls include compliance with the applicable portions of the FDA’s Quality System Regulation (“QSR”), which sets forth good manufacturing practice requirements; facility registration and product reporting of adverse medical events listing; truthful and non-misleading labeling; and promotion of the device only for its cleared or approved intended uses. Class II devices are also subject to these general controls, and any other special controls as deemed necessary by the FDA to ensure the safety and effectiveness of the device. Review and clearance by the FDA for these devices is typically accomplished through the so-called 510(k) pre-market notification procedure. A Class III device requires approval of a premarket application (“PMA”), an expensive, lengthy and uncertain process requiring many years to complete.
When 510(k) clearance is sought, a sponsor must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to a previously approved device. If the FDA agrees that the proposed device is substantially equivalent to the predicate device, then 510(k) clearance to market will be granted. After a device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, requires a new 510(k) clearance or could require pre-market approval. Our instruments and disposables used for the production of cell cultures are generally regulated as Class I devices exempt from the 510(k) clearance process.
Clinical trials are almost always required to support a PMA and are sometimes required for a 510(k) pre-market notification. These clinical trials generally require submission of an application for an investigational device exemption (“IDE”). An IDE must be supported by pre-clinical data, such as animal and laboratory testing results, which show that the device is safe to test in humans and that the study protocols are scientifically sound. The IDE must be approved in advance by the FDA for a specified number of patients, unless the product is deemed a non-significant risk device and is eligible for more abbreviated IDE requirements.
Both before and after a medical device is commercially distributed, manufacturers and marketers of the device have ongoing responsibilities under FDA regulations. The FDA reviews design and manufacturing practices, labeling and record keeping, and manufacturers’ required reports of adverse experiences and other information to identify potential problems with marketed medical devices. Device manufacturers are subject to periodic and unannounced inspection by the FDA for compliance with the QSR, cGMP requirements that govern the methods used in, and the facilities and controls used for, the design, manufacture, packaging, servicing, labeling, storage, installation, and distribution of all finished medical devices intended for human use.
If the FDA finds that a manufacturer has failed to comply or that a medical device is ineffective or poses an unreasonable health risk, it can institute or seek a wide variety of enforcement actions and remedies, ranging from a public warning letter to more severe actions such as: fines, injunctions, and civil penalties; recall or seizure of products; operating restrictions, partial suspension or total shutdown of production; refusing requests for 510(k) clearance or PMA approval of new products; withdrawing 510(k) clearance or PMA approvals already granted; and criminal prosecution.
The FDA also has the authority to require repair, replacement or refund of the cost of any medical device.
The FDA also administers certain controls over the export of medical devices from the U.S., as international sales of medical devices that have not received FDA approval are subject to FDA export requirements. Additionally, each foreign country subjects such medical devices to its own regulatory requirements. In the EU, a single regulatory approval process has been created, and approval is represented by the “CE” Mark.
Other Regulation in the United States
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (2010) (collectively, the “PPACA”). Enacted in March 2010, the PPACA, makes changes that could significantly impact the development and commercialization and of our product candidates. Significant measures contained in the PPACA include, for example, coordination and promotion of research on comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. The PPACA also includes significant new fraud and abuse measures, including required disclosures of financial arrangements with physician customers, lower thresholds for violations and increasing potential penalties for such violations. In addition, the PPACA establishes an Independent Payment Advisory Board (“IPAB”), to reduce the per capita rate of growth in Medicare spending. The IPAB has broad discretion to propose policies to reduce expenditures, which may have a negative impact on payment rates for services or therapeutics, including therapeutics like BiovaxID. In addition to the PPACA, the effect of which cannot presently be fully quantified given its relatively recent enactment, various healthcare reform proposals have also emerged from federal and state governments. Changes in healthcare policy could substantially impact the development and commercialization of BiovaxID. We cannot predict whether future healthcare initiatives will be implemented at the federal or state level or in countries outside of the U.S. in which we may do business, or the effect any future legislation or regulation will have on us. The taxes imposed by the new federal legislation and the expansion in government's role in the U.S. healthcare industry may result in decreased profits to us, lower reimbursements by payers for our product candidates or reduced medical procedure volumes, all of which may adversely affect our business, financial condition and results of operations, possibly materially.
The Biologics Price Competition and Innovation Act (2010). The Biologics Price Competition and Innovation Act, establishes an abbreviated approval pathway for “biosimilar” biological products. Among the provisions potentially applicable to our product candidates are (1) innovator manufacturers of reference biological products (such as BiovaxID) are granted 12 years of exclusive use before biosimilars can be approved for marketing in the U.S. and (2) an application for a biosimilar product may not be submitted to the FDA until 4 years after the date on which the BLA for the reference product was first approved. FDA is still early in the process of developing regulations to implement the provisions of this legislation.
Toxic Substances Control Act. The EPA has promulgated regulations under Section 5 of the Toxic Substances Control Act (“TSCA”), which require notification procedures for review of certain so-called intergeneric microorganisms before they are introduced into commerce. Intergeneric microorganisms are those formed by deliberate combinations of genetic material from organisms classified in different taxonomic genera, which are types of animal or plant groups. The regulations provide exemptions from the reporting requirements for new microorganisms used for research and development when the researcher or institution is in mandatory compliance with the National Institutes of Health Guidelines for Research Involving Recombinant DNA Molecules (“NIH Guidelines”). Those researchers voluntarily following the NIH Guidelines can, by documenting their use of the NIH Guidelines, satisfy EPA’s requirements for testing in contained structures. The EPA may enforce the TSCA through enforcement actions such as seizing noncompliant substances, seeking injunctive relief, and assessing civil or criminal penalties. We believe that our research and development activities involving intergeneric microorganisms comply with the TSCA, but there can be no assurance that restrictions, fines or penalties will not be imposed on us in the future.
Health Care Coverage and Reimbursement. Commercial success in marketing and selling our product candidates depends, in part, on the availability of adequate coverage and reimbursement from third-party health care payers, such as government and private health insurers and managed care organizations. Third-party payers are increasingly challenging the pricing of medical products and services. Government and private sector initiatives to limit the growth of health care costs, including price regulation, competitive pricing, coverage and payment policies, and managed-care arrangements, are continuing in many countries where we do business, including the U.S. These changes are causing the marketplace to put increased emphasis on the delivery of more cost-effective medical products.
Government programs, including Medicare and Medicaid, private health care insurance and managed-care plans have attempted to control costs by limiting the amount of reimbursement they will pay for particular procedures or treatments. This has created an increasing level of price sensitivity among customers for our product candidates. Examples of how limits on drug coverage and reimbursement in the U.S. may cause drug price sensitivity include the growth of managed care, changing Medicare reimbursement methodologies, and drug rebates and price controls. Some third-party payers must also approve coverage for new or innovative devices or therapies before they will reimburse health care providers who use the medical devices or therapies. Even though a new medical product may have been cleared for commercial distribution, we may find limited demand for the product until reimbursement approval has been obtained from governmental and private third-party payers.
Anti-Kickback Laws. In the U.S., there are federal and state anti-kickback laws that prohibit the payment or receipt of kickbacks, bribes or other remuneration to induce the purchase, order or recommendation of health care products and services. These laws constrain the sales, marketing and other promotional activities of pharmaceutical companies, such as us, by limiting the kinds of financial arrangements we may have with prescribers, purchasers, dispensers and users of drugs and biologics. The U.S. Department of Health and Human Services (“HHS”) Office of Inspector General (“OIG”) has issued “Compliance Guidance” for pharmaceutical manufacturers which, among other things, identifies manufacturer practices implicating the federal anti-kickback law (42 U.S.C. §1320a-7b(b)) and describes elements of an effective compliance program. The OIG Compliance Guidance is voluntary, and we have not adopted a formal compliance program modeled after the one described in the OIG Compliance Guidance. Although none of our practices have been subject to challenge under any anti-kickback laws, due to the breadth of the statutory provisions of some of these laws, it is possible that some of our practices might be challenged under one or more of these laws in the future. Violations of these laws can lead to civil and criminal penalties, including imprisonment, fines and exclusion from participation in federal health care programs. Any such violations could have a material adverse effect on our business, financial condition, results of operations or cash flows.
Health Information Privacy and Security. Individually identifiable health information is subject to an array of federal and state regulation. Federal rules promulgated pursuant to the Health Information Portability and Accountability Act of 1996 (“HIPAA”) regulate the use and disclosure of health information by “covered entities” (which includes individual and institutional providers from which we may receive individually identifiable health information). These regulations govern, among other things, the use and disclosure of health information for research purposes, and require the covered entity to obtain the written authorization of the individual before using or disclosing health information for research. Failure of the covered entity to obtain such authorization (absent obtaining a waiver of the authorization requirement from an Institutional Review Board) could subject the covered entity to civil and criminal penalties. As the implementation of this regulation is still in its early phases, we may experience delays and complex negotiations as we deal with each entity’s differing interpretation of the regulations and what is required for compliance. Further, HIPAA’s criminal provisions are not limited in their applicability to “covered persons,” but apply to any “person” that knowingly and in violation of the statute obtains or discloses individually identifiable health information. Also, where its customers or contractors are covered entities, including hospitals, universities, physicians or clinics, we may be required by the HIPAA regulations to enter into “business associate” agreements that subject us to certain privacy and security requirements, including making its books and records available for audit and inspection by HHS and implementing certain health information privacy and security safeguards. In addition, many states have laws that apply to the use and disclosure of health information, and these laws could also affect the manner in which we conduct its research and other aspects of its business. Such state laws are not preempted by the federal privacy law where they afford greater privacy protection to the individual. While activities to assure compliance with health information privacy laws are a routine business practice, we are unable to predict the extent to which its resources may be diverted in the event of an investigation or enforcement action with respect to such laws.
Foreign Regulation
Whether or not we obtain FDA approval for a product, we may choose to seek approval of our product candidates by the comparable regulatory authorities of foreign countries. Like in the U.S., before we can commence clinical trials or marketing of our product candidates in a foreign country, we must submit a new drug application in that country. The requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country. Although governed by the applicable country, clinical trials conducted outside of the U.S. typically are administered under a three-phase sequential process similar to that discussed above for pharmaceutical products. Clinical trials conducted in the EU must comply with the EU Clinical Trial Regulations and are monitored and inspected by each EU Member State. Clinical trials conducted in Canada must comply with the Health Canada Regulations and are approved and monitored by an independent committee of doctors, scientists, advocates and others to ensure safety and ethical standards. In addition, regulatory approval of pricing/marketing of the product candidate is required in most countries other than the U.S. We face the risk that the pricing, which results from the regulatory approval process, would be insufficient to generate an acceptable return to us or our collaborators.
The regulatory/marketing approval process varies from country to country, and the approval time may be longer or shorter than that required for FDA approval. However, foreign countries are similar in that they generally require, as part of any new drug submission/application process, sufficient evidence to support the safety, efficacy and quality of the product candidate.
Under EU regulatory systems, the manufacture and sale of new drugs are controlled by the EMA. MAAs are submitted under either a centralized or decentralized procedure for most products. The centralized procedure, which is the pathway for BiovaxID, is available for medicines produced by biotechnology or which are highly innovative and provides single marketing authorization that is valid for all EU member states. Under European Commission Regulation 726/2004, the centralized authorization procedure is required for all biotechnology-derived medicinal products developed through recombinant DNA technology, controlled expression of genes coding for biologically active proteins, and hybridoma and monoclonal antibody methods. It is also required for designated orphan medicinal products and all new active substances indicated for the treatment of AIDS, cancer, neurodegenerative disorder, or diabetes. Under the EMA centralized procedure, the marketing approval of BiovaxID can be simultaneously obtained throughout all EU-member countries with a single MAA. As part of the EMA’s centralized procedure, the MAA is assessed by the EMA’s CHMP, which designates from within its membership, a Rapporteur and Co-Rapporteur, as well as a PRAC Rapporteur and Co-Rapporteur. The Rapporteurs and Co-Rapporteurs are assigned with the primary responsibility of preparing and delivering an approvability evaluation report, supported by a team of assessors from their National Authority. On December 3, 2013, we submitted an MAA under a centralized procedure with the EMA for BiovaxID.
Under Canadian regulatory systems, the manufacture and sale of new drugs are controlled by Health Canada. NDSs are be submitted for regulatory approval in Canada. Upon sufficient evidence to support safety, efficacy or quality claims for a NDS, a product is issued a Notice of Compliance (“NOC”) and a Drug Identification Number (“DIN”) indicating that the biologic is approved for sale in Canada. All drugs that are marketed in Canada are subject to the Food and Drugs Act and Regulations. The Biologics and Genetic Therapies Directorate is responsible for the review and approval of all types of drug submissions for Biological (Schedule D) and Radiopharmaceutical (Schedule C) drug products, including, but not limited to NDSs. For the regulatory requirements specific to Biological (Schedule D to the Act) drugs, please refer to Divisions 4, of Part C of the Regulations. For general regulatory requirements concerning all drugs, including Good Manufacturing Practices, please refer to Divisions 1, 1a, 2, 5 and 8 of Part C of the Regulations.
Third-Party Reimbursement and Pricing Controls
In the U.S. and in international markets, sales of pharmaceutical products depend in significant part on the availability of reimbursement to the consumer from third-party payers, such as government and private insurance plans. Third-party payers are increasingly challenging the prices charged for medical products and services. It will be time-consuming and expensive for us to go through the process of seeking reimbursement from Medicare and private payers. Our product candidates may not be considered cost effective, and coverage and reimbursement may not be available or sufficient to allow us to sell our product candidates on a competitive and profitable basis. The passage of the Medicare Prescription Drug and Modernization Act of 2003 imposes new requirements for the distribution and pricing of prescription drugs which may affect the marketing of our product candidates.
In many foreign markets, including the member states of the EU, pricing of pharmaceutical products is subject to governmental control, mainly through pricing and reimbursement regulations under the centralized healthcare systems in place in EU member states. In some EU jurisdictions, an approved medication cannot be marketed until an agreed-upon price structure is in place. Various forms of pricing controls tend to exert downward pressure on pricing of pharmaceutical products in the EU. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
Liability Insurance
We may be exposed to potential product liability claims by users of our product candidates. Our business segments may expose us to potential risk of liability. We seek to obtain agreements from contract production customers to mitigate such potential liability and to indemnify us under certain circumstances. The terms and conditions of our sales and instruments include provisions which are intended to limit our liability for indirect, special, incidental or consequential damages. There can be no assurance, however, that we will be successful in obtaining such agreements or that such indemnification, if obtained, will adequately protect us against potential claims. The terms and conditions of our sales and instruments include provisions which are intended to limit our liability for indirect, special, incidental or consequential damages. We presently maintain product liability insurance coverage in the aggregate and per occurrence of $2.0 million, in connection with our product candidates and our products and services, in amounts which we believe to be adequate and on acceptable terms.
Although, we believe that our current level of coverage is adequate to protect our business from foreseeable product liability claims, we may seek to increase our insurance coverage in the future in the event that we significantly increase our level of contract product and services and/or the initiation of any clinical trial program. There can be no assurance; however, that we will be able to maintain our existing coverage or obtain additional coverage on acceptable terms, or that such insurance will provide adequate coverage against all potential claims to which we may be exposed. A successful partially or completely uninsured claim against us could have a material adverse effect on our operations.