UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form
| (Mark One) | ||
| | ||
| OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Quarterly Period Ended
or
| OF THE SECURITIES EXCHANGE ACT OF 1934 | ||
|
For the Transition Period from ___________ to ___________ |
Commission File Number:
Filana Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| | | ||
| (State or other jurisdiction of | (I.R.S. Employer | ||
| incorporation or organization) | Identification Number) |
(
(Address, including zip code, of registrant’s principal executive offices and
telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | Trading Symbol(s) | Name of each exchange on which registered | ||
| | | |
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer ☐ | Accelerated Filer ☐ | |
| | Smaller Reporting Company | |
| Emerging Growth Company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes
Indicate the number of shares outstanding of each of the issuer’s classes of common stock, as of the latest practicable date.
| Common Stock, $0.001 par value | | ||
| Shares Outstanding as of May 4, 2026 |
TABLE OF CONTENTS
| Page No. |
||
| PART I. |
FINANCIAL INFORMATION |
|
| Item 1. |
||
| Condensed Consolidated Balance Sheets – March 31, 2026 and December 31, 2025 |
||
| Condensed Consolidated Statements of Operations – Three Months Ended March 31, 2026 and 2025 |
||
| Condensed Consolidated Statements of Cash Flows – Three Months Ended March 31, 2026 and 2025 |
||
| Item 2. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
|
| Item 3. |
||
| Item 4. |
||
| PART II. |
OTHER INFORMATION |
|
| Item 1. |
||
| Item 1A |
||
| Item 2. |
||
| Item 3. |
||
| Item 4. |
||
| Item 5. |
||
| Item 6. |
||
PART I. FINANCIAL INFORMATION
Item 1. Financial Statements
CONDENSED CONSOLIDATED BALANCE SHEETS
(Unaudited, In thousands, except share and par value data)
| March 31, 2026 | December 31, 2025 | |||||||
| ASSETS | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Prepaid expenses and other current assets | ||||||||
| Total current assets | ||||||||
| Property and equipment, net | ||||||||
| Total assets | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS' EQUITY | ||||||||
| Current liabilities: | ||||||||
| Accounts payable and accrued expenses | $ | $ | ||||||
| Accrued development expense | ||||||||
| Accrued compensation and benefits | ||||||||
| Other current liabilities | ||||||||
| Total current liabilities | ||||||||
| Other non-current liabilities | ||||||||
| Total liabilities | ||||||||
| Commitments and contingencies (Notes 8, 9 and 10) | ||||||||
| Stockholders' equity: | ||||||||
| Preferred stock, $ par value; shares authorized, issued and outstanding | ||||||||
| Common stock, $ par value; shares authorized; shares issued and outstanding at March 31, 2026 and December 31, 2025 | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated deficit | ( | ) | ( | ) | ||||
| Total stockholders' equity | ||||||||
| Total liabilities and stockholders' equity | $ | $ | ||||||
See accompanying notes to condensed consolidated financial statements.
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS
(Unaudited, in thousands, except per share data)
| Three months ended |
||||||||
| March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Operating expenses: |
||||||||
| Research and development |
$ | $ | ||||||
| General and administrative |
||||||||
| Total operating expenses |
||||||||
| Operating loss |
( |
) | ( |
) | ||||
| Interest income |
||||||||
| Other income (loss), net |
( |
) | ||||||
| Net loss |
$ | ( |
) | $ | ( |
) | ||
| Net loss per share, basic and diluted |
$ | ( |
) | $ | ( |
) | ||
See accompanying notes to condensed consolidated financial statements.
CONDENSED CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(Unaudited, in thousands, except share data)
| Total |
||||||||||||||||||||
| Common stock |
Additional |
Accumulated |
stockholders' |
|||||||||||||||||
| Shares |
Par value |
paid-in capital |
deficit |
equity |
||||||||||||||||
| Balance at December 31, 2024 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
| Stock-based compensation for: |
||||||||||||||||||||
| Stock options for employees |
— | |||||||||||||||||||
| Stock options for non-employees |
— | |||||||||||||||||||
| Issuance of common stock pursuant to exercise of stock options |
||||||||||||||||||||
| Net income |
— | ( |
) | ( |
) | |||||||||||||||
| Balance at March 31, 2025 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
| Balance at December 31, 2025 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
| Stock-based compensation for: |
||||||||||||||||||||
| Stock options for employees |
— | |||||||||||||||||||
| Stock options for non-employees |
— | |||||||||||||||||||
| Net loss |
— | ( |
) | ( |
) | |||||||||||||||
| Balance at March 31, 2026 |
$ | $ | $ | ( |
) | $ | ||||||||||||||
See accompanying notes to condensed consolidated financial statements.
CONDENSED CONSOLIDATED STATEMENTS OF CASH FLOWS
(Unaudited, in thousands)
| Three months ended March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Cash flows from operating activities: |
||||||||
| Net loss |
$ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||
| Stock-based compensation |
||||||||
| Depreciation and amortization |
||||||||
| Changes in operating assets and liabilities: |
||||||||
| Prepaid and other current assets |
||||||||
| Accounts payable and accrued expenses |
( |
) | ||||||
| Accrued development expense |
||||||||
| Accrued compensation and benefits |
( |
) | ( |
) | ||||
| Other liabilities |
( |
) | ( |
) | ||||
| Net cash used in operating activities |
( |
) | ( |
) | ||||
| Cash flows from financing activities: |
||||||||
| 2025 ATM program offering costs |
( |
) | ||||||
| Proceeds from issuance of common stock upon exercise of stock options |
||||||||
| Net cash (used in) provided by financing activities |
( |
) | ||||||
| Net decrease in cash and cash equivalents |
( |
) | ( |
) | ||||
| Cash and cash equivalents at beginning of period |
||||||||
| Cash and cash equivalents at end of period |
$ | $ | ||||||
See accompanying notes to condensed consolidated financial statements.
Notes to Condensed Consolidated Financial Statements
(Unaudited)
Note 1. General and Liquidity
Filana Therapeutics, Inc. and its wholly-owned subsidiary (collectively referred to as the “Company”) discovers and develops proprietary pharmaceutical product candidates that may offer significant improvements to patients and healthcare professionals. The Company currently focuses its discovery and product development efforts on disorders of the nervous system.
The accompanying unaudited condensed consolidated financial statements of the Company have been prepared in accordance with accounting principles generally accepted in the United States (“GAAP”) for interim financial information and pursuant to the instructions to the Quarterly Report on Form 10-Q and Article 10 of Regulation S-X. All intercompany transactions and balances have been eliminated in consolidation. Accordingly, the condensed consolidated financial statements do not include all of the information and footnotes required by GAAP for complete consolidated financial statements. In the opinion of management of the Company, all adjustments, consisting of normal recurring adjustments, considered necessary for a fair presentation have been included. Operating results for the three months ended March 31, 2026 are not necessarily indicative of the results that may be expected for any other interim period or for the year 2026. For further information, refer to the consolidated financial statements and footnotes thereto included in the Company’s Annual Report on Form 10-K for the year ended December 31, 2025.
Liquidity
The Company has incurred significant net losses and negative cash flows since inception, and as a result has an accumulated deficit of $
Note 2. Significant Accounting Policies
Use of Estimates
The Company makes estimates and assumptions in preparing its condensed consolidated financial statements in conformity with GAAP. These estimates and assumptions affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements and the reported amount of expenses incurred during the reporting period. The Company evaluates its estimates on an ongoing basis, including those estimates related to legal liabilities, stock-based compensation, clinical trials and manufacturing agreements. Actual results could differ from these estimates and assumptions.
Cash and Cash Equivalents and Concentration of Credit Risk
The Company invests in cash and cash equivalents. The Company considers highly liquid financial instruments with original maturities of three months or less to be cash equivalents. Highly liquid investments that are considered cash equivalents include money market accounts and funds, certificates of deposit, and U.S. Treasury securities. The Company maintains its cash and cash equivalents at one financial institution.
Fair Value Measurements
The Company recognizes financial instruments in accordance with the authoritative guidance on fair value measurements and disclosures for financial assets and liabilities. This guidance defines fair value, establishes a framework for measuring fair value in accordance with GAAP, and expands disclosures about fair value measurements. The guidance also establishes a three-tier fair value hierarchy, which prioritizes the inputs used in measuring fair value. These tiers include:
| ● | Level 1 includes quoted prices in active markets. |
| ● | Level 2 includes significant observable inputs, such as quoted prices for identical or similar securities, or other inputs that are observable and can be corroborated by observable market data for similar securities. The Company uses market pricing and other observable market inputs obtained from third-party providers. It uses the bid price to establish fair value where a bid price is available. The Company does not have any financial instruments where the fair value is based on Level 2 inputs. |
| ● | Level 3 includes unobservable inputs that are supported by little or no market activity. The Company does not have any financial instruments where the fair value is based on Level 3 inputs. |
If a financial instrument uses inputs that fall in different levels of the hierarchy, the instrument will be categorized based upon the lowest level of input that is significant to the fair value calculation. The fair value of cash and cash equivalents was based on Level 1 inputs at March 31, 2026 and December 31, 2025.
Business Segments
The Company reports segment information based on how it internally evaluates the operating performance of its business units, or segments. The Company’s operations are confined to business segment: the development of novel drugs and diagnostics.
The Company’s reportable segment reflects the manner in which its chief operating decision maker (“CODM”) allocates resources and assesses performance. The Company's CODM is the President and Chief Executive Officer. The primary measure used by the Company's CODM for purposes of allocating resources is based on net loss that also is reported on the consolidated statements of operations as consolidated net loss. The measure of segment assets is reported on the condensed consolidated balance sheets as cash and cash equivalents.
The Company has not generated any product revenue in the current period and expects to continue to incur significant expenses and operating losses for the foreseeable future as we advance our product candidates through stages of development and clinical trials.
As such, the CODM uses cash forecast models in deciding how to invest in the development of novel drugs and diagnostics. Such cash forecast models are reviewed to assess the entity-wide operating results and performance. Net loss is used to monitor budget versus actual results. Monitoring budgeted versus actual results, net cash used in operating activities for the period and cash on hand are used in assessing performance of the segment.
The following table summarizes expenses by category regularly reviewed by the CODM (in thousands):
| Three months ended | ||||||||
| March 31, | ||||||||
| 2026 | 2025 | |||||||
| Operating expenses | ||||||||
| Research and development | $ | ( | ) | $ | ( | ) | ||
| General and administrative | ( | ) | ( | ) | ||||
| Other segment items(a) | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
(a) Other segment items include interest income and other income (loss), net.
The following table summarizes assets regularly reviewed by the CODM (in thousands):
| March 31, 2026 | December 31, 2025 | |||||||
| Cash and cash equivalents | $ | $ | ||||||
For the three months ended March 31, 2026 and 2025, the net cash used in operating activities was $
Stock-based Compensation
The Company recognizes non-cash expense for the fair value of all stock options and other share-based awards. The Company uses the Black-Scholes option valuation model to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. This model requires the input of subjective assumptions including expected stock price volatility, expected life and estimated forfeitures of each award. These assumptions consist of estimates of future market conditions, which are inherently uncertain, and therefore, are subject to management's judgment. For all options granted, it recognizes the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally to years.
The Company has granted share-based awards that vest upon achievement of certain performance criteria (“Performance Awards”). The Company multiplies the number of Performance Awards by the fair value of its common stock on the date of grant to calculate the fair value of each award. It estimates an implicit service period for achieving performance criteria for each award. The Company recognizes the resulting fair value as expense over the implicit service period when it concludes that achieving the performance criteria is probable. It periodically reviews and updates as appropriate its estimates of implicit service periods and conclusions on achieving the performance criteria. Performance Awards vest and common stock is issued upon achievement of the performance criteria.
Net Loss per Share
The Company computes basic net loss per share on the basis of the weighted-average number of common shares outstanding for the reporting period. Diluted net loss per share is computed on the basis of the weighted-average number of common shares outstanding plus potential dilutive common shares outstanding using the treasury-stock method. Potential dilutive common shares consist of outstanding common stock options and Performance Awards. There is no difference between the Company’s net loss and comprehensive loss. The numerators and denominators in the calculation of basic and diluted net loss per share were as follows (in thousands, except net loss per share data):
| Three months ended | ||||||||
| March 31, | ||||||||
| 2026 | 2025 | |||||||
| Numerator: | ||||||||
| Net loss | $ | ( | ) | $ | ( | ) | ||
| Denominator: | ||||||||
| Shares used in computing net loss per share, basic and diluted | ||||||||
| Net loss per share, basic and diluted | $ | ( | ) | $ | ( | ) | ||
| Dilutive common stock options excluded from net loss per share, diluted | ||||||||
| Dilutive Performance Awards excluded from net loss per share, diluted | ||||||||
The Company excluded all common stock options and Performance Awards outstanding for the three months ended March 31, 2026 and 2025 from the calculation of net loss per share, diluted, because the effect of including outstanding options and Performance Awards would have been anti-dilutive.
Fair Value of Financial Instruments
Financial instruments include accounts payable, accrued expenses, accrued development expense and other liabilities. The estimated fair value of certain financial instruments may be determined using available market information or other appropriate valuation methodologies. However, considerable judgment is required in interpreting market data to develop estimates of fair value; therefore, the estimates are not necessarily indicative of the amounts that could be realized or would be paid in a current market exchange. The effect of using different market assumptions and/or estimation methodologies may be material to the estimated fair value amounts. The carrying amounts of accounts payable, accrued expenses, accrued development expense and other liabilities are at cost, which approximates fair value due to the short maturity of those instruments.
Research Contracts, Prepaids and Accruals
The Company has entered into various research and development contracts with research institutions and other third-party vendors. These agreements are generally cancelable. Related payments are recorded as research and development expenses as incurred. The Company records prepaids and accruals for estimated ongoing research costs. When evaluating the adequacy of prepaid expenses and accrued liabilities, the Company analyzes progress of the studies including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates are made in determining the prepaid and accrued balances at the end of any reporting period. Actual results could differ from the Company’s estimates. The Company’s historical prepaid and accrual estimates have not been materially different from actual costs.
Incentive Bonus Plan
In 2020, the Company established the 2020 Cash Incentive Bonus Plan (the “CIB Plan”) to incentivize CIB Plan participants. Awards under the CIB Plan are accounted for as liability awards under ASC 718 “Stock-based Compensation”. The fair value of each potential CIB Plan award will be determined once a grant date occurs and will be remeasured each reporting period. Compensation expense associated with the CIB Plan will be recognized over the expected achievement period for each CIB Plan award, when a Performance Condition (as defined below) is considered probable of being met. See Note 9 for further discussion of the CIB Plan.
Property and Equipment
Property and equipment are recorded at cost, net of accumulated depreciation. Depreciation is recorded using the straight-line method over the estimated useful lives of the assets. Owned buildings and related improvements have estimated useful lives of
Property and equipment are reviewed for impairment when events or changes in circumstances indicate the carrying amount of an asset may not be recoverable. If property and equipment are considered to be impaired, an impairment loss is recognized.
Income Taxes
The Company accounts for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax balances are adjusted to reflect tax rates based on currently enacted tax laws, which will be in effect in the years in which the temporary differences are expected to reverse. The Company has accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of certain deferred tax assets is dependent upon future earnings. The Company is uncertain about the timing and amount of any future earnings. Accordingly, the Company offsets these deferred tax assets with a valuation allowance.
The Company accounts for uncertain tax positions in accordance with ASC 740, “Income Taxes”, which clarifies the accounting for uncertainty in tax positions. These provisions require recognition of the impact of a tax position in the Company’s condensed consolidated financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected as a component of income tax expense.
Recently Issued Accounting Pronouncements
In November 2024, the FASB issued ASU 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40): Disaggregation of Income Statement Expenses. This ASU includes amendments that require entities to bifurcate specified expense line items on the income statement into underlying components, including employee compensation, depreciation, and intangible asset amortization, as applicable. Qualitative descriptions of the remaining components are required. These enhanced disclosures are required for both interim and annual periods. In January 2025, the FASB subsequently issued ASU 2025-01, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (Subtopic 220-40): Clarifying the Effective Date, to provide clarification on the ASU's effective date. The new standard is effective for fiscal years beginning after December 15, 2026 on a prospective basis with the option to apply it retrospectively, and for interim periods within fiscal years beginning after December 15, 2027. Early adoption is permitted. The adoption of this guidance will result in the Company being required to include enhanced disclosures around income statement expenses.
Note 3. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets at March 31, 2026 and December 31, 2025 consisted of the following (in thousands):
| March 31, |
December 31, |
|||||||
| 2026 |
2025 |
|||||||
| Prepaid insurance |
$ | $ | ||||||
| Contract research organization and other deposits |
||||||||
| Interest receivable |
||||||||
| Other |
||||||||
| Total prepaid expenses and other current assets |
$ | $ | ||||||
Contract research organization and other deposits represent cash payments made to vendors in excess of expenses incurred.
Note 4. Real Property and Other Income, Expense
The Company owns a two-building office complex in Austin, Texas, a portion of which serves as its corporate headquarters. Maintenance, physical facilities, leasing, property management and other key responsibilities related to property ownership are outsourced to professional real-estate managers. The office complex has approximately
The Company records the net income (loss) from building operations and leases as other income (loss), net, as leasing is not core to the Company’s operations. Building depreciation and amortization for space not occupied by the Company is included in general and administrative expense. Building depreciation and amortization for space occupied by the Company is allocated between general and administrative expense and research and development expense. Components of other income (loss), net, for the periods presented were as follows (in thousands):
| Three months ended |
||||||||
| March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Lease revenue |
$ | $ | ||||||
| Property operating expenses |
( |
) | ( |
) | ||||
| Other income (loss), net |
$ | $ | ( |
) | ||||
The Company had accrued property taxes related to the building totaling $
Gross rental income under existing third-party leases at March 31, 2026 is as follows (in millions):
| For the year ending December 31, |
||||
| 2026 |
||||
| 2027 |
||||
| 2028 |
||||
| 2029 |
||||
| 2030 and beyond |
||||
| Total gross rental income |
Note 5. Property and equipment
The components of property and equipment, net, as of March 31, 2026 and December 31, 2025 were as follows (in thousands):
| March 31, | December 31, | |||||||
| 2026 | 2025 | |||||||
| Land | $ | $ | ||||||
| Buildings | ||||||||
| Site improvements | ||||||||
| Tenant improvements | ||||||||
| Furniture and equipment | ||||||||
| Construction in progress | ||||||||
| Other | ||||||||
| Gross property and equipment | $ | $ | ||||||
| Accumulated depreciation | ( | ) | ( | ) | ||||
| Property and equipment, net | $ | $ | ||||||
Depreciation expense for property and equipment was $
Note 6. Stockholders’ Equity and Stock-Based Compensation Expense
Stock Option and Performance Award Activity in 2026
During the three months ended March 31, 2026, stock options and unvested Performance Awards outstanding under the Company’s stock option plans changed as follows:
| Stock Options |
Performance Awards |
|||||||
| Outstanding as of December 31, 2025 |
||||||||
| Options granted |
||||||||
| Options exercised |
||||||||
| Options forfeited/canceled |
||||||||
| Outstanding as of March 31, 2026 |
||||||||
The weighted average exercise price per share of options outstanding at March 31, 2026 was $
During the three months ended March 31, 2026, there were stock options exercised.
Stock-based Compensation Expense in 2026
During the three months ended March 31, 2026 and 2025, the Company’s stock-based compensation expense was as follows (in thousands):
| Three months ended |
||||||||
| March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Research and development |
$ | $ | ||||||
| General and administrative |
||||||||
| Total stock-based compensation expense |
$ | $ | ||||||
The Company estimates forfeitures for each award in determining stock-based compensation expense. There were
2018 Equity Incentive Plan
The Company’s Board of Directors (the “Board”) or a designated committee of the Board is responsible for administration of the Company’s 2018 Omnibus Incentive Plan, as amended in May 2022 (the “2018 Plan”) and determines the terms and conditions of each option granted, consistent with the terms of the 2018 Plan. The Company’s employees, directors, and consultants are eligible to receive awards under the 2018 Plan, including grants of stock options and Performance Awards. Share-based awards generally expire years from the date of grant. The 2018 Plan provides for issuance of up to
When stock options or Performance Awards are exercised net of the exercise price and taxes, the number of shares of stock issued is reduced by the number of shares equal to the amount of taxes owed by the award recipient and that number of shares are cancelled. The Company then uses its cash to pay tax authorities the amount of statutory taxes owed by and on behalf of the award recipient.
Note 7. Income Taxes
The Company did provide for income taxes during the three months ended March 31, 2026 because it has projected a taxable net loss for the full year 2026 for which any benefit will be offset by an increase in the valuation allowance. There was also provision for income taxes for the three months ended March 31, 2025.
Note 8. Commitments
The Company conducts its product research and development programs through a combination of internal and collaborative programs that include, among others, arrangements with universities, contract research organizations and clinical research sites. The Company has contractual arrangements with these organizations that are cancelable. The Company’s obligations under these contracts are largely based on services performed. The Company also had non-cancellable commitments for the manufacture of simufilam's active pharmaceutical ingredient totaling approximately $
On February 26, 2025, the Company entered into a License Agreement (the “License Agreement”) with Yale University (“Yale”) pursuant to which the Company was granted exclusive worldwide rights, with rights to sublicense, to Yale’s interest in certain patent and other intellectual property rights that could be useful or necessary to the development and commercialization of simufilam for the treatment of Tuberous Sclerosis Complex (TSC)-related epilepsy and other potential indications. Pursuant to the License Agreement, the Company has agreed to use reasonable commercial efforts to implement a plan that it has designed for such development and commercialization. In exchange for the rights acquired pursuant to the License Agreement, the Company paid Yale (i) a nominal upfront license fee, and agreed to pay (ii) payments upon the achievement of specified clinical, regulatory and commercial milestones, totaling up to $
Unless earlier terminated, the License Agreement will continue on a country-by-country basis until the later of (i) the date on which the last valid claim of the license patents expires or otherwise lapses, (ii) the end of any government or regulatory exclusivity period, and (iii) 10 years following the date of first sale of licensed product in such country.
Note 9. 2020 Cash Incentive Bonus Plan
In August 2020, the Board approved the CIB Plan. The CIB Plan was subsequently amended, as described below on March 6, 2025. The CIB Plan was established as an “at-risk” cash bonus program that would reward CIB Plan participants with additional cash compensation in lockstep with significant increases in the Company’s market valuation (as measured by market capitalization or potential merger consideration). The CIB Plan is considered “at-risk” because CIB Plan participants will not receive a cash bonus unless the Company’s market valuation increases significantly and certain other conditions specified in the CIB Plan are met. Specifically, CIB Plan participants will not be paid any cash bonuses unless (1) the Company completes a merger or acquisition transaction that constitutes a sale of ownership of the Company or its assets (a Merger Transaction) or (2) the Compensation Committee of the Board (the Compensation Committee) determines the Company has sufficient cash on hand, as defined in the CIB Plan. CIB Plan participants will be paid all earned cash bonuses allocated under the CIB Plan in the event of a Merger Transaction.
Because of the inherent discretion and uncertainty regarding the CIB Plan requirements, the Company has concluded that a CIB Plan grant date has not occurred as of March 31, 2026.
The Company’s market valuation for purposes of the CIB Plan is determined based on either (1) the closing price of one share of the Company's common stock on the Nasdaq Capital Market multiplied by the total issued and outstanding shares and options to purchase shares of the Company, or (2) the aggregate consideration payable to security holders of the Company in a Merger Transaction. This constitutes a market condition under applicable accounting guidance.
The CIB Plan (as amended March 6, 2025) triggers a potential cash bonus each time the Company’s market valuation increases significantly, up to a maximum $
Payment of cash bonuses is deferred until such time as (1) the Company completes a Merger Transaction, or (2) the Compensation Committee determines the Company has sufficient cash on hand to render payment (each, a “Performance Condition”), neither of which may ever occur. Accordingly, CIB Plan participants may never be paid a cash bonus that is awarded under the CIB Plan, even if the Company’s market capitalization increases significantly.
The CIB Plan is accounted for as a liability award. The fair value of each Valuation Milestone award will be determined once a grant date occurs and will be remeasured each reporting period. Compensation expense associated with the CIB Plan will be recognized over the expected achievement period for each of the Valuation Milestones, when a Performance Condition is considered probable of being met.
In October 2020, the Company achieved the first Valuation Milestone based on market capitalization. Subsequently in 2020, the Compensation Committee approved a potential cash bonus award of $
During the year ended December 31, 2021, the Company achieved
On February 13, 2025, the Compensation Committee concluded that
As discussed in Note 10, on January 24, 2025, the Delaware Court of Chancery entered a Final Order and Judgment approving the Stipulation and dismissing an action related to the CIB Plan with prejudice. The Final Order and Judgment required the Company to further amend the CIB Plan, as provided in the Stipulation, which was completed on March 6, 2025. The March 6, 2025 Amendment specifies that upon the occurrence of a Merger Transaction, the Chairman, President and CEO (assuming such person holds all three such offices) will not be entitled to any payments in respect of any Market Capitalization Conditions, but will instead be entitled to receive a bonus payment based upon the Merger Transaction only. Cash bonuses to the Chairman, President and CEO (assuming such person holds all three offices) are not subject to discretionary allocation under the CIB Plan. No person currently holds all three offices of Chairman, President and CEO.
Note 10. Contingencies
The Company is, and from time to time, the Company may become, involved in litigation or other legal proceedings and claims, including U.S. government inquiries, investigations and Citizen Petitions submitted to the U.S. Food and Drug Administration (“FDA”). In addition, the Company has received, and from time to time may receive, inquiries from government authorities relating to matters arising from the ordinary course of business. The outcome of these proceedings is inherently uncertain. Regardless of outcome, legal proceedings can have an adverse impact on the Company because of defense and settlement costs, diversion of management resources, and other factors. At this time, no assessment can be made as to their likely outcome or whether the outcome will be material to the Company other than as disclosed below. The Company believes that its total provisions for legal matters are adequate based upon currently available information.
Government Investigations
Beginning in August 2021, the Company received subpoenas and other requests for documents and information from the Department of Justice Fraud Section (“DOJ”), document requests from the SEC, and a Civil Investigative Demand from the Civil Division of the Department of Justice. These requests sought corporate information and documents concerning the research and development of simufilam and/or SavaDx, and the Company cooperated fully with each.
On September 26, 2024, the Company announced that it had reached a settlement with the SEC resolving the SEC investigation of the Company’s disclosures regarding its Phase 2b clinical trial of simufilam for the treatment of Alzheimer’s disease (the “Phase 2b Study”) and related matters. The SEC also agreed to a settlement with two former senior employees of the Company. Pursuant to these settlements, the U.S. District Court for the Western District of Texas (the “Texas District Court”) entered final consent judgment on October 18, 2024, on a complaint filed by the SEC against the Company and its two former senior employees. The Company has neither admitted nor denied the allegations of the complaint. The SEC’s complaint alleged that certain disclosures by the Company regarding the Phase 2b Study violated certain federal securities laws and SEC rules, including negligence-based disclosure violations of Sections 17(a)(2) and 17(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”), as well as recordkeeping and reporting requirements under the Securities Exchange Act of 1934, as amended (the “Exchange Act”). The complaint alleges that the Company’s SEC reports and other public statements regarding the Phase 2b Study negligently contained materially misleading statements and omissions. The SEC’s allegations with respect to the Company’s two former employees relate to these employees’ roles in such disclosures. Under its settlement, the Company consented to a permanent injunction against future violations of Section 17(a) of the Securities Act, Section 13(a)(1) of the Exchange Act and Rules 12b-20, 13a-1, 13a-11 and 13a-13 under the Exchange Act. In addition, the Company paid a civil monetary penalty of $40 million in November 2024.
On February 18, 2026, the DOJ notified the Company that it had closed its inquiry into the Company regarding allegations of research misconduct as described in the indictment in United States v. Wang, 8:24-cr-000211-TDC (D. Md.). As previously disclosed, that indictment was dismissed with prejudice by DOJ on October 23, 2025.
These outcomes end the previously disclosed investigations of the Company by the DOJ and SEC.
Securities Class Actions and Shareholder Derivative Actions
Between August 27, 2021 and October 26, 2021, four putative class action lawsuits were filed alleging violations of the federal securities laws by the Company and certain named officers. The complaints rely on allegations contained in Citizen Petitions submitted to FDA and allege that various statements made by the defendants regarding simufilam were rendered materially false and misleading. The Citizen Petitions were all subsequently denied by FDA. These actions were filed in the Texas District Court. The complaints seek unspecified compensatory damages and other relief on behalf of a purported class of purchasers of the Company’s securities.
On June 30, 2022, a federal judge consolidated the class action lawsuits into case (the “Consolidated Securities Action”) and appointed a lead plaintiff and a lead counsel. Lead plaintiff filed a consolidated amended complaint on August 18, 2022 on behalf of a putative class of purchasers of the Company’s securities between September 14, 2020 and July 26, 2022. On May 11, 2023, the Texas District Court dismissed with prejudice plaintiffs’ claims against defendant Nadav Friedmann, PhD, MD, our former Chief Medical Officer and a Company director, who is now deceased, but otherwise denied defendants’ motion to dismiss. Defendants filed an answer to the consolidated amended complaint on July 3, 2023. On February 22, 2024, plaintiffs filed a motion to supplement their complaint to extend the putative class period through October 12, 2023. The Texas District Court granted that Motion on June 12, 2024, and plaintiffs filed a supplemental complaint on June 13, 2024.
On February 2, 2024, a putative class action lawsuit was filed alleging violations of the federal securities law by the Company and certain named officers. The complaint relies on an October 12, 2023 article that describes a purported leaked report of alleged scientific misconduct by a scientific collaborator of the Company at City University of New York (the “CUNY Article”). The complaint alleges that various statements made by the defendants regarding simufilam were rendered materially false and misleading by this article. The action was filed in the U.S. District Court for the Northern District of Illinois (the “Illinois District Court”). The complaint seeks unspecified compensatory damages and other relief on behalf of a purported class of purchasers of the Company’s securities between August 18, 2022 and October 12, 2023. On May 28, 2024, the Illinois District Court transferred this action to the Texas District Court, where it was consolidated into the Consolidated Securities Action.
On November 13, 2024, Plaintiffs in the Consolidated Securities Action filed a second motion to supplement their complaint. On March 13, 2024, plaintiffs filed a Motion for Class Certification. On February 25, 2025, the Texas District Court denied Plaintiff’s motion for class certification without prejudice, explaining that rulings on pending motions, including specifically Plaintiffs’ second motion to supplement their complaint, could affect disposition of class certification. On May 21, 2025, the Texas District Court granted Plaintiffs’ second motion for leave to file a second supplemented complaint and instructed Plaintiffs to refile their motion for class certification. Plaintiffs filed their second supplemental complaint on May 22, 2025, and reasserted their motion for class certification on June 20, 2025. The Texas District Court granted Plaintiffs’ motion for class certification on August 12, 2025. On October 21, 2025, the United States Court of Appeals for the Fifth Circuit agreed to hear Defendants’ interlocutory appeal challenging the District Court order granting class certification. Briefing was completed on April 20, 2026.
The parties to the Consolidated Securities Action engaged in mediation with David M. Murphy of Phillips Alternative Dispute Resolutions Enterprises (the “Mediator”) beginning in May 2025. On December 19, 2025, plaintiffs in the Consolidated Securities Action filed a Final Alternative Dispute Resolution (“ADR”) Report with the Texas District Court as required by Local Rule CV-88(g). As stated in the Final ADR Report, the Company and one of its officers (the “Settling Defendants”) and plaintiffs have accepted a double-blind Mediator’s Recommendation, reflected in a binding term sheet (the “Term Sheet”) pursuant to which the Settling Defendants will pay plaintiffs $
The Term Sheet provides that the Company will pay a settlement amount equal to $
The Settling Defendants and plaintiffs intend to prepare a formal stipulation of settlement and motion for preliminary approval of the settlement describing the terms of the proposed settlement, which will be presented to the Texas District Court for preliminary approval. Following preliminary approval of the proposed settlement by the Texas District Court and a notice and review period for class members, plaintiffs will seek final approval of the proposed settlement from the Texas District Court.
The Company recorded a loss contingency of $
On November 4, 2021, a shareholder derivative action related to the initial four class action lawsuits filed in the Texas District Court was filed, purportedly on behalf of the Company, in the Texas District Court, asserting claims under the U.S. securities laws and state fiduciary duty laws against certain named officers and the members of the Company’s Board. This complaint relies on the allegations made in Citizen Petitions that were submitted to (and subsequently denied by) FDA. The complaint alleges, among other things, that the individual defendants exposed the Company to unspecified damages and securities law liability by causing it to make materially false and misleading statements, in violation of the U.S. securities laws and in breach of their fiduciary duties to the Company. The derivative case seeks, among other things, to recover unspecified compensatory damages on behalf of the Company arising out of the individual defendants’ alleged wrongful conduct. Although the plaintiff in this derivative case does not seek relief against the Company, the Company has certain indemnification obligations to the individual defendants. Between November 4, 2021 and November 9, 2023,
Beginning on March 18, 2024, shareholder derivative actions related to the February 2024 class action lawsuit originally filed in the Illinois District Court were filed, purportedly on behalf of the Company, in the Illinois District Court, asserting claims under the U.S. securities laws and state fiduciary duty laws against certain named officers and the members of the Company’s Board. The complaints rely on the CUNY Article. The complaints allege, among other things, that the individual defendants exposed the Company to unspecified damages and securities law liability by causing it to make materially false and misleading statements, in violation of the U.S. securities laws and in breach of their fiduciary duties to the Company. The derivative cases seek, among other things, to recover unspecified compensatory damages on behalf of the Company arising out of the individual defendant’s alleged wrongful conduct. Although the plaintiffs in these derivative cases do not seek relief against the Company, the Company has certain indemnification obligations to the individual defendants. On September 6, 2024, these cases were consolidated and stayed pending further developments in the shareholder class action initially filed in the Illinois District Court on February 2, 2024. Due to the stage of the foregoing shareholder derivative actions, the Company is unable to predict the outcome or estimate the amount of loss or range of losses that could potentially result from such derivative lawsuits.
On December 13, 2024, a putative class action lawsuit (the “2024 Securities Class Action”) was filed, purportedly on behalf of the Company, alleging violations of the federal securities law by the Company and certain named officers. The complaint alleged that various statements made by the defendants regarding simufilam were revealed to be materially false and misleading by the release of top-line results for the Company’s RETHINK-ALZ Phase 3 clinical trial on November 25, 2024. The 2024 Securities Class Action was filed in the Texas District Court. The complaint sought unspecified compensatory damages and other relief on behalf of a purported class of purchasers of the Company’s securities between February 7, 2024, and November 24, 2024.
On May 8, 2025, the Court appointed a lead plaintiff and lead counsel for the 2024 Securities Class Action. Lead plaintiff filed an amended complaint on August 25, 2025, on behalf of a putative class of purchasers of the Company’s securities between October 13, 2023 and March 25, 2025. The amended complaint added new officer defendants and additional allegations that various statements made by the defendants regarding simufilam were revealed to be materially false and misleading by disclosures in June 2024 through September 2024. On September 19, 2025, defendants filed a motion to consolidate the 2024 Securities Class Action into the Consolidated Securities Action. Lead plaintiff opposed that motion, and it remains pending.
On November 6, 2025, Defendants moved to dismiss the 2024 Securities Class Action. Lead plaintiff opposed that motion, and it remains pending.
Due to the stage and procedural posture of the 2024 Securities Class Action, the Company is unable to predict the likelihood of an unfavorable outcome in the 2024 Securities Class Action. It is reasonably possible, however, that the Company may incur a loss. The Company is unable to reasonably estimate the amount or range of potential loss at this time.
Subject to the Term Sheet in the Consolidated Securities Action, the Company intends to defend all pending shareholder derivative actions and securities class actions vigorously.
Anti-SLAPP Lawsuit
On August 6, 2024, a lawsuit was filed in the District Court for the Southern District of New York which, as amended and as joined by intervenor plaintiffs, asserts claims, including a claim under the New York Anti-SLAPP Law, against the Company and against former officers to whom the Company has certain indemnification obligations. The amended complaint and the complaint in intervention seeks costs and damages relating to a defamation action filed by the Company against the plaintiffs and subsequently dismissed voluntarily and without prejudice by the Company. The Company and its two former officers named as defendants have entered comprehensive settlement agreements with the intervenor plaintiffs and another potential plaintiff under which the parties provided mutual releases and the Company paid approximately $
Litigation Contingencies
In connection with ongoing litigation, the Company has reserved an amount of $
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with Filana Therapeutics, Inc.’s (the “Company,” “we,” “us,” or “our”) condensed consolidated financial statements and accompanying notes included elsewhere in this Quarterly Report on Form 10-Q. Operating results are not necessarily indicative of results that may occur in future periods.
Forward-looking Statements and Notices
This Quarterly Report on Form 10-Q contains certain statements that are considered forward-looking statements. Forward-looking statements relate to expectations, beliefs, projections, future plans and strategies, anticipated events or trends and similar expressions concerning matters that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “should,” “will” and “would” or the negatives of these terms or other comparable terminology.
The forward-looking statements are based on our beliefs, assumptions and expectations of our future performance, taking into account all information currently available to us. Forward-looking statements involve risks and uncertainties and our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Examples of such forward-looking statements include, but are not limited to statements about:
| ● | the timing and conduct of our planned and future potential clinical trials and studies for Simufilam in Tuberous Sclerosis Complex (TSC) and additional indications, including the timing of initiation, enrollment and completion of the trials and the period during which the results of the trials will become available; | |
| ● |
the safety profile or treatment benefits, if any, of simufilam; |
|
| ● |
our ability to conduct planned preclinical studies of simufilam relating to epilepsy in TSC; | |
| ● | our ability to initiate, conduct or analyze additional clinical and non-clinical studies with our product candidates targeted at central nervous system disorders, including TSC-related epilepsy; | |
| ● | our ability to successfully carry out our obligations under our license agreement with Yale, for which we have assumed all responsibility for global development and commercialization for simufilam for the treatment of TSC-related epilepsy; | |
| ● | our plans or ability to expand therapeutic indications for simufilam outside of TSC-related epilepsy; | |
| ● | our reliance on third-party contractors to conduct all of our clinical and non-clinical trials and to make drug supply, or their ability to do so on-time or on-budget; | |
| ● |
the interpretation of data from our pre-clinical or early clinical studies, such as Phase 1 and Phase 2 studies, and any studies that are not randomized controlled trials; |
|
| ● | the impact of pre-clinical findings on our ability to develop our product candidates; | |
| ● | our use of exploratory ‘research use only’ non-safety related biomarkers in our clinical studies; | |
| ● |
our ability to file for and obtain regulatory approval of our product candidates; |
|
| ● |
our strategy and ability to establish an infrastructure to commercialize any product candidates, if approved; |
|
| ● |
the potential future revenues of our product candidates, if approved and commercialized; |
|
| ● |
the market acceptance of our product candidates, if approved and commercialized; |
|
| ● |
the pricing and reimbursement of our product candidates, if approved and commercialized; |
|
| ● |
the utility of protection, or the sufficiency, of our intellectual property; |
|
| ● |
our potential competitors or competitive products for therapeutic areas we may elect to pursue; | |
| ● |
our need to raise new capital from time to time to finance our operations and the impact of macroeconomic conditions on our ability to effectively raise capital; |
|
| ● | expectations regarding trade secrets, technological innovations, licensing agreements and outsourcing of certain business functions; | |
| ● |
our expenses or incurred costs increasing by material amounts in excess of budgeted amounts due to unexpected cost overruns, imperfect forecasting, increased scope of activities or other causes; | |
| ● |
fluctuations in our financial or operating results; | |
| ● |
our operating losses, anticipated operating and capital expenditures and legal expenses; | |
| ● |
expectations regarding the issuance of shares of common stock, options or other equity to employees or directors pursuant to equity compensation awards, net of employment taxes; | |
| ● |
the development and maintenance of our internal information systems and infrastructure; |
| ● | our ability to minimize the likelihood and impact of adverse cybersecurity incidents in our information systems and infrastructure; | |
| ● |
our ability to attract and retain personnel; |
|
| ● |
existing or emerging regulations and regulatory developments in the United States and other jurisdictions in which we operate; |
|
| ● |
our expectations regarding the appropriate size and scope of our operations; |
|
| ● |
the sufficiency of our cash resources to continue to fund our operations; |
|
| ● |
potential future agreements with third parties in connection with the commercialization of our product candidates; |
|
| ● |
the accuracy of our estimates regarding expenses, loss contingency reserves, capital requirements, and needs for additional financing; |
|
| ● |
assumptions and estimates used for our disclosures regarding stock-based compensation; |
|
| ● |
expense, timing and outcome of pending or future litigation or other legal proceedings and claims (including U.S. government inquiries) including the potential for negotiated settlements of such matters; and | |
| ● |
litigation, claims or other uncertainties that may arise from allegations made against us or our former employees or collaborators and the potential resolution of same. |
Please also refer to the section entitled “Risk Factors” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2025 and this Quarterly Report on Form 10-Q, as such risk factors may be further amended, updated or modified periodically in our reports filed with the U.S. Securities and Exchange Commission (the “SEC”) for further information on these and other risks affecting us.
We caution you not to place undue reliance on forward-looking statements because our future results may differ materially from those expressed or implied by them. We do not undertake to update any forward-looking statement, whether written or oral, relating to the matters discussed in this Quarterly Report on Form 10-Q, except as required by law.
This Quarterly Report on Form 10-Q may also contain statistical data and drug information received from our independent consultants or based on industry publications or other publicly available information. We have not independently verified the accuracy or completeness of the data contained in these sources of data and information. Accordingly, we make no representations as to the accuracy or completeness of such data and information. You are cautioned not to give undue weight to such data and information.
Our research programs in neurodegeneration have historically benefited from scientific and financial support from the National Institutes of Health (“NIH”). The contents of this Quarterly Report on Form 10-Q are solely our responsibility and do not necessarily represent any official views of NIH, the Department of Health and Human Services, or any other agency of the United States government, or any of our vendors, collaborators or unrelated third-parties.
All our pharmaceutical assets under development are investigational product candidates. These have not been approved for use in any medical indication by any regulatory authority in any jurisdiction and their safety, efficacy or other desirable attributes, if any, have not been established in any patient population. Consequently, none of our product candidates are approved or available for sale anywhere in the world.
Our clinical results from earlier-stage clinical trials may not be indicative of future results from later-stage or larger scale clinical trials and do not ensure regulatory approval. You are cautioned that subsequent results may differ materially.
Overview
Filana Therapeutics, Inc. is a clinical-stage biotechnology company based in Austin, Texas. Our mission is to develop transformative medicines to improve the lives of patients with central nervous system (CNS) disorders, such as Tuberous Sclerosis Complex (TSC)-related epilepsy and other diseases associated with dysregulation or overexpression of filamin A. Our science is based on modulating the activity of a critical protein in the brain for patients with certain CNS disorders, such as TSC. Our lead therapeutic drug candidate, simufilam, was under clinical evaluation for the proposed treatment of Alzheimer’s disease in two Phase 3 clinical studies through 2024, when all ongoing clinical trials for simufilam in Alzheimer’s disease were discontinued. The phase out of the Company’s Alzheimer's disease development program was completed in the second quarter of 2025.
Our lead therapeutic product candidate, simufilam, is a proprietary small molecule oral treatment drug being studied for the treatment of TSC-related epilepsy. Simufilam was discovered and designed in-house and was characterized by our academic collaborators during research activities that were conducted from approximately 2008 to date.
Simufilam is intended to modulate the activity of a protein called filamin A, which regulates diverse aspects of neuronal cell development. Our and our collaborators’ published studies have demonstrated that the combination of mTOR activation and filamin A overexpression causes cell overgrowth, abnormal connectivity, and brain malformations, leading to seizures in patients with TSC. While such seizures have historically been treated with mTOR inhibitors and other drugs, filamin A represents a novel target for the potential treatment of TSC-related epilepsy, independent of mTOR. Simufilam is a potential first-in-class filamin A-modulating agent with the potential to treat TSC-related epilepsy. We continue to explore other potential indications for simufilam, both CNS related and otherwise, where our pre-clinical experiments suggest that simufilam may provide a benefit.
We Own or Control Worldwide Rights to Our Drug Development Program
Our success depends in part on our ability to obtain, maintain, and enforce intellectual property protection for our product candidates, including simufilam, and related technologies. We seek to protect our proprietary position through a combination of patents, patent applications, trade secrets, know‑how, and other intellectual property rights.
We own or exclusively control a portfolio of issued patents and pending patent applications directed to simufilam and related diagnostic assets. These intellectual property rights include patents covering composition of matter for simufilam in the United States, Europe, Australia, Israel, and Canada, as well as patents covering certain methods of use of simufilam in the United States, Europe, and Japan. We also own pending patent applications covering diagnostic assets related to simufilam in the United States, Europe, Japan, China, Canada, and Australia, as well as additional pending patent applications covering other diagnostic assets in the United States, Europe, Japan, China, and Canada.
In the United States, our patent protection relating to simufilam, including its solid forms and methods of use for Alzheimer’s disease and other neurodegenerative diseases, consists of nine issued U.S. patents, with expiration dates ranging from 2029 to 2040, subject to potential patent term extensions and adjustments. We have filed corresponding foreign patent applications for each of these U.S. patent families where we believe such protection is commercially appropriate.
In addition to our owned intellectual property, on February 26, 2025, we entered into a License Agreement (the “License Agreement”) with Yale University (“Yale”) pursuant to which we were granted exclusive worldwide rights, with rights to sublicense, to Yale’s interest in certain patent and other intellectual property rights that could be useful or necessary to the development and commercialization of simufilam for the treatment of TSC‑related epilepsy and other potential indications. Under this exclusive, worldwide license, we have the right to sublicense the licensed intellectual property. The license is subject to customary diligence obligations and includes milestone payments, royalties on net sales of licensed products, and other contingent payments, including payments tied to the transfer of any regulatory priority review voucher, if issued. As a result, commercial sales of simufilam or related products, if any, may be subject to royalty obligations to Yale.
Except for intellectual property licensed from third parties, including pursuant to the License Agreement with Yale, we do not owe royalties or other payments to third parties with respect to our owned patents and patent applications covering simufilam. For more information, please also refer to the section entitled “––License Agreement with Yale.”
About Tuberous Sclerosis Complex (TSC)
TSC is a genetic disorder that results from a mutation in the TSC1 or TSC2 genes. According to the Tuberous Sclerosis Alliance (TSCA), TSC is estimated to affect around 1 in 6,000 live births with approximately 50,000 people affected in the United States and more than one million worldwide. Clinical symptoms of TSC are variable and can impact organ systems with non-malignant tumors developing in brain, eyes, heart, kidney, skin and lungs. Interrelated neuropsychiatric manifestations of TSC such as intellectual disability, autism spectrum disorder, anxiety, aggression, attention deficit hyperactivity disorder, and sleep disturbance can significantly impact quality of life. Epilepsy is common in TSC, occurring in 84% of patients registered in the TSC Alliance Natural History Database with onset often occurring in their first year of life. Approximately 60 percent of TSC patients suffer from treatment-resistant seizures despite use of multiple anti-seizure medications. TSC-related epilepsy is associated with poor outcomes and increases the risk of cognitive deficits.
Preclinical Studies with Simufilam in Tuberous Sclerosis Complex
Preclinical research conducted at Yale indicates that simufilam (then PTI-125) may be effective in reducing TSC-related seizure activity. A study conducted by Angelique Bordey, PhD, at Yale and published in the peer-reviewed journal Neuron in 2014 found overexpression of filamin A and neuronal abnormalities in brain tissue from TSC patients, as well as in brain tissue from mice that had been genetically altered to model TSC. The study further found that genetically normalizing filamin A expression in the mice prevented neuronal abnormalities. The paper, MEK-ERK1/2-Dependent FLNA Overexpression Promotes Abnormal Dendritic Patterning in Tuberous Sclerosis Independent of mTOR (Neuron. 2014 Oct 1. PMID: 25277454), was co-authored by Zhang L, Bartley CM, Gong X, Hsieh LS, Lin TV and Feliciano DM.
A later study conducted by Dr. Bordey and published in the peer-reviewed journal Science Translational Medicine in 2020 likewise found elevated filamin A and neuronal abnormalities in brain tissue from patients with focal cortical dysplasia type II (FCDII), a genetic disorder with similar characteristics to TSC, as well as in the brains of mice that had been genetically altered to model TSC and FCDII. That study showed that normalizing filamin A expression in the mice through genetic knockdown both limited the neuronal abnormalities and reduced the frequency of epileptic seizures in the mice. This 2020 paper, Filamin A Inhibition Reduces Seizure Activity in a Mouse Model of Focal Cortical Malformations (Sci Transl Med. 2020 Feb 19. PMID: 32076941), was co-authored by Zhang L, Huang T, Teaw S, Nguyen LH, Hsieh LS, Gong X, and Lindsay Burns, a former employee of the Company.
In this 2020 study, Dr. Bordey and her colleagues also studied the effect of treatment with simufilam in the mice that had been altered to model TSC and FCDII. The research showed that treatment with simufilam limited neuronal abnormalities and reduced seizure activity in the mouse model at a level similar to genetic knockdown of filamin A expression.
Dr. Bordey joined the Company as Senior Vice President, Neuroscience, on May 1, 2025, while continuing her tenured academic position at Yale School of Medicine on a part time basis. In connection with plans to initiate a clinical program in TSC-related epilepsy, the Company appointed Dr. Joseph Hulihan as Chief Medical Officer in August 2025. Dr. Hulihan, who brings over 25 years of industry experience, devotes approximately half of his professional time to the Company and will advise on the clinical development of simufilam.
In August 2025, the Company announced positive preclinical results of a study evaluating simufilam in a well-accepted mouse model of TSC-related epilepsy. The study was conducted in collaboration with the TSCA and the TSC Preclinical Consortium using an animal model of TSC-related epilepsy, the Tsc1 conditional knockout (CKO) mouse line (Tsc1-CKO). These mice develop spontaneous seizures and are used by the TSC Alliance to evaluate the effectiveness and safety of novel and repurposed therapeutics in the potential treatment of TSC-related epilepsy. The study was conducted by PsychoGenics, Inc., the TSC Preclinical Consortium’s research partner.
The Tsc1-CKO mice were treated with several doses of simufilam. Seizure activity was monitored for approximately three weeks after onset, and simufilam was evaluated against treatment with vehicle alone. The data showed that simufilam attenuated the progression of seizure activity, with a statistically significant correlation between simufilam dose and the number of seizures by the end of the study. Not all parameters measured reached statistical significance.
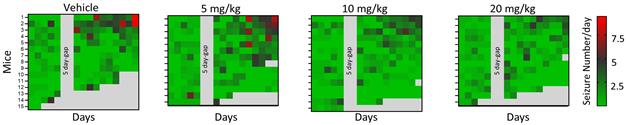
Data and analyses were published in Epilepsia on April 14, 2026. Figure 3C from the article is a heatmap comparing the number of seizures per day recorded for each mouse in the study. Mice were administered vehicle (control) or simufilam at one of three ascending doses.
Epilepsia paper, Figure 3C
Heatmap of the daily number of seizures per mouse during electroencephalography (EEG) recordings in each condition. EEG monitoring spanned 20 days, but seizure counts were quantified for the first 5 and last 10 days, and comparative analysis was performed between the first and last 5 days. Gray squares: Mice had died. The 5 day-gap indicates that analysis was not performed for this period.
Status of Investigational New Drug (IND) Application
Based on the results of these studies, the Company filed an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) in order to initiate a proof-of-concept clinical trial for simufilam in TSC-related epilepsy. On December 15, 2025, the Company received a formal letter from FDA confirming that the proposed clinical trial is on full clinical hold subject to the Company providing FDA with additional information, including additional pre-clinical data, and modifying the protocol design.
The Company is working expeditiously to address the items identified in the letter. However, as announced on December 18, 2025, the Company no longer expects to initiate a proof-of-concept clinical trial for simufilam in TSC-related epilepsy in the first half of 2026. The updated timing for initiation of a clinical trial will depend on the Company’s ability to provide the requested information to FDA and on satisfactory completion of FDA’s review.
License Agreement with Yale
On February 26, 2025, we entered into the License Agreement with Yale pursuant to which we were granted exclusive worldwide rights, with rights to sublicense, to Yale’s interest in certain patent and other intellectual property rights that could be useful or necessary to the development and commercialization of simufilam for the treatment of TSC-related epilepsy and other potential indications. Pursuant to the License Agreement, we have agreed to use reasonable commercial efforts to implement a plan designed for such development and commercialization.
In exchange for the rights acquired pursuant to the License Agreement, we paid Yale (i) a nominal upfront license fee, and agreed to pay (ii) payments upon the achievement of specified clinical, regulatory and commercial milestones, totaling up to $4.5 million and (iii) upon transfer to a third party of a regulatory priority review voucher, if issued, a low-to-mid double digit percentage of any consideration received for such transfer. We also agreed to pay Yale tiered royalties, ranging from a low- to mid- single digit percentage, on aggregate net sales of licensed products, subject to tiered minimum annual royalty payments ranging from the low- to mid- hundreds of thousands of dollars.
Unless earlier terminated, the License Agreement will continue on a country-by-country basis until the later of (i) the date on which the last valid claim of the license patents expires or otherwise lapses, (ii) the end of any government or regulatory exclusivity period, and (iii) 10 years following the date of first sale of licensed product in such country. We may terminate the License Agreement: (i) at our option, upon specified advance notice to Yale and (ii) if Yale commits a material breach of the License Agreement that is not cured within a specified timeframe. Yale may terminate the License Agreement under specified circumstances, including if we (i) fail to make any payment due under the License Agreement and fail to cure such non-payment within a specified timeframe, (ii) commit a breach of the License Agreement that is not cured within a specified timeframe, (iii) default on a material obligation to any creditor, unless cured within a specified timeframe, (iv) fail to obtain or maintain insurance required by the License Agreement, (v) bring or assist a patent challenge against Yale (or if a sublicensee does so). In addition, the License Agreement will terminate automatically upon the occurrence of certain bankruptcy and insolvency events involving us.
The License Agreement includes customary confidentiality, reporting and inspection, and indemnification provisions.
Safety Data From our Now-Discontinued Alzheimer’s Disease Program
Prior to discontinuing the Alzheimer’s disease program in early 2025, we conducted several Phase 1, Phase 2 and Phase 3 trials of simufilam in the indication. During this clinical trial program, more than 2,000 patients were treated with simufilam, with some patients receiving treatment for as long as 24 months. The safety observations for simufilam from the clinical trials in the discontinued Alzheimer's disease program remain relevant and important to research and clinical trials in pursuit of other indications, including TSC-related epilepsy.
Phase 1 Clinical Safety Study in Healthy Volunteers
The FDA accepted our IND submission for simufilam in 2017. This IND included the requisite preclinical studies concerning safety pharmacology, toxicology, genotoxicity and bioanalytical methods.
We thereafter investigated the safety, dosing and pharmacokinetic profile of simufilam in healthy human volunteers. The design of our first-in-human Phase 1 study was based on regulatory feedback, clinical and scientific rationale and observations from previously conducted preclinical and in vitro studies. In this Phase 1 study, simufilam was evaluated in 24 healthy human volunteers (18 simufilam, 6 placebo) in a single site in the U.S. for safety, tolerability and pharmacokinetics. Study subjects were administered a single oral dose of 50, 100 or 200 mg of simufilam or placebo. Drug appeared safe and well-tolerated. Importantly, simufilam showed no treatment-related adverse effects and no dose-limiting safety findings. Pharmacokinetic measurements demonstrated that simufilam, a small molecule, was rapidly absorbed. Dose-proportionality was observed over the full dose range of 50 to 200 mg.
Phase 2 24-Month Clinical Safety Study of Simufilam in Alzheimer’s Disease
In March 2020, we initiated a Phase 2 clinical safety study of simufilam in patients with Alzheimer’s disease (NCT04388254). This study was funded in part by a research grant award from NIH. This study was designed to evaluate the long-term clinical safety and tolerability of simufilam in patients with Alzheimer’s disease over 24 months. This study enrolled over 200 patients with Alzheimer’s disease who were recruited from 16 U.S. clinical sites.
We conducted the 24-month safety study in three continuous phases:
| ● |
a 12-month, open-label treatment phase, followed by |
|
| ● |
a 6-month randomized, placebo-controlled withdrawal phase, followed by |
|
| ● |
6 additional months of open-label treatment. |
Study participants received simufilam oral tablets 100 mg twice-daily in the open-label treatment phases, and simufilam or matching placebo during the randomized withdrawal phase. In an open-label study design, both the health providers and the patients are aware of the drug treatment being given.
All study participants who completed 12 months of open-label simufilam treatment were eligible to participate in the 6-month randomized, placebo-controlled withdrawal phase. Likewise, all study participants who completed the randomized, placebo-controlled withdrawal phase were eligible for 6 additional months of open-label treatment.
During this 24-month study, oral simufilam 100 mg tablets twice daily appeared safe and well tolerated. No drug-related serious adverse events were observed. The most common treatment-emergent adverse events (TEAEs) were Covid-19 and urinary tract infection.
Phase 3 Clinical Trials of Simufilam in Alzheimer’s Disease
Between 2021 and 2025 we enrolled 1,900 patients—including 1,153 patients treated with simufilam during the blinded phase—in two randomized placebo-controlled Phase 3 clinical trials of oral simufilam in mild-to-moderate Alzheimer’s disease. The 800 patients enrolled in the first Phase 3 study, called RETHINK-ALZ, received either 100 mg simufilam tablets or placebo twice daily for 52 weeks (NCT04994483). The 1,125 patients enrolled in the second Phase 3 study, called REFOCUS-ALZ, received either 100 mg and 50 mg simufilam tablets or placebo twice daily for 76 weeks (NCT05026177). Patients who completed either of these Phase 3 studies or the 24-month Phase 2 safety study were eligible to enter a 12-month Open Label Extension study in which all patients received simufilam.
On November 25, 2024, we announced that RETHINK-ALZ did not meet each of the pre-specified co-primary, secondary and exploratory biomarker endpoints. We thereafter discontinued the Phase 3 REFOCUS-ALZ study and Open Label Extension study. On March 25, 2025, we announced that REFOCUS-ALZ also did not meet each of the pre-specified co-primary, secondary and exploratory biomarker endpoints. With the failure of both trials to meet their efficacy endpoints, we announced the decision to phase out our Alzheimer’s disease development program. That process was completed in the second quarter of 2025.
Throughout both of these Phase 3 trials and the subsequent open-label extension study simufilam demonstrated a favorable safety profile. The drug’s adverse event profile was comparable to placebo, and no drug-related serious adverse events were observed.
Peer Reviewed Article Describing the Results of our Phase 3 Trials of Simufilam in Alzheimer’s Disease
On January 13, 2026, we announced the publication of the article Phase 3 randomized clinical trials of simufilam in mild-to-moderate Alzheimer’s disease in the Journal of Prevention of Alzheimer’s Disease (JPAD). The paper provides a detailed analysis of data from the RETHINK-ALZ and REFOCUS-ALZ clinical trials. Although, the two studies did not meet their pre-specified efficacy endpoints, exploratory post-hoc analysis of the studies contained in this article offered informative insights regarding simufilam.
Risk is Fundamental to the Drug Development Process
We are in the business of new drug discovery and development. Our research and development activities are long, complex, costly and involve a high degree of risk. Holders of our common stock should carefully read our 2025 Annual Report on Form 10-K and any subsequent Quarterly and Current Reports filed with the SEC in their entirety, including important disclosures under the caption “Risk Factors.” Because risk is fundamental to the process of drug discovery and development, you are cautioned to not invest in our publicly traded securities unless you are prepared to sustain a total loss of the money you have invested.
Our Science
Our Science is Based on Modulating the Activity of Filamin A
Our scientific approach is to treat central nervous system diseases by modulating the activity of the filamin A scaffolding protein. Based on existing research, we believe that filamin A is a promising target for drug development with respect to TSC-related epilepsy.
Research that we sponsored has identified a family of high-affinity, small molecules to target filamin A as a means of potentially treating central nervous system disorders such as TSC-related epilepsy. This family of small molecules, including simufilam, our lead small molecule (oral) therapeutic product candidate, was designed in-house and characterized by our academic collaborators.
Filamin A is a scaffolding protein found in the brain and elsewhere in the body. Scaffolding proteins bring multiple proteins together, coordinating their interaction. Our product candidate, simufilam, is a potential first-in-class filamin A-modulating agent with the potential to treat TSC-related epilepsy. We continue to explore other potential indications for simufilam, both CNS related and otherwise, where our pre-clinical experiments suggest that simufilam may provide a benefit.
Published research shows that patients with TSC have elevated levels of filamin A and that this overexpression of filamin A, in combination with mTOR activation, causes cell overgrowth, abnormal connectivity, and brain malformations, leading to seizures. Preclinical studies in a mouse model showed that genetically normalizing filamin A levels reduced seizure activity and limited neuronal abnormalities independent of mTOR activation.
In this same mouse model, treatment with simufilam reduced seizure activity and limited neuronal abnormalities in a manner similar to genetically normalizing filamin A levels. A second study in a well-accepted mouse model of TSC-related epilepsy showed that treatment with simufilam attenuated the progression of seizure activity.
Extension of Our Science to Other Indications
We may leverage our scientific insights in neurodegeneration and neuroinflammation and advanced tools in molecular biology, biochemistry, and imaging to expand our science to other diseases, including other diseases in which filamin A may be associated with the disease process. New indications and new drug development approaches may complement or supersede our current focus priorities.
In addition, we are exploring and incorporating artificial intelligence (AI) capabilities and related data analytics to target improvements in productivity and efficiency in our business, enhancements to research and development activities, and advancements in statistical analysis capabilities.
Simufilam Drug Supply
We have a drug supply agreement with Evonik Industries AG for simufilam. Under the agreement, Evonik supplies and is expected to continue to supply us with clinical-grade quantities of simufilam. Evonik is one of the world’s largest contract development and manufacturing organizations for pharmaceutical ingredients. Other vendors supply excipients, the finished dosage form (i.e., simufilam tablets), drug packaging, package labeling and other critical components of the supply chain for drug supply that can be used for clinical studies.
SavaDx
Our product candidate, called SavaDx, was an early-stage program focused on detecting the presence of Alzheimer’s disease from a small sample of blood. For business, technical and personnel reasons, we discontinued the development of SavaDx as of mid-2025. Development activity related to SavaDx accounted for less than 1% of our research budget in all periods presented prior to the program’s discontinuation.
Financial Overview
We have yet to generate any revenues from product sales. We have an accumulated deficit of $506.4 million at March 31, 2026. These losses have resulted principally from costs incurred in connection with research and development activities, salaries and other personnel-related costs, legal related costs and general corporate expenses. Research and development activities include costs of clinical and preclinical trials as well as clinical supplies associated with our product candidates. Salaries and other personnel-related costs include stock-based compensation associated with stock options and other equity awards granted to employees and non-employees. Our operating results may fluctuate substantially from period to period as a result of enrollment rates of clinical trials for our product candidates, timing of preclinical activities and our need for clinical supplies.
We expect to continue to use significant cash resources in our operations for the next several years. Our cash requirements for operating activities and capital expenditures may increase in the future as we:
| ● | engage with, and satisfactorily respond to, requests for additional information from the FDA concerning the clinical hold on our IND application for simufilam in TSC-related epilepsy | |
| ● |
conduct preclinical and clinical studies for our product candidates, including simufilam for TSC-related epilepsy; | |
| ● |
seek regulatory approvals for our product candidates; |
|
| ● |
develop, formulate, manufacture and commercialize our product candidates; |
|
| ● |
acquire or in-license additional products or technologies, or expand the use of our technology; |
|
| ● |
maintain, defend and expand the scope of our intellectual property; and |
|
| ● |
expend resources related to legal proceedings and claims, including U.S. government inquiries. |
Product revenue will depend on our ability to receive regulatory approvals for, and successfully market, our product candidates. If our development efforts result in regulatory approval and successful commercialization of our product candidates, we expect to generate revenue from direct sales of our drugs and/or, if we license our drugs to future collaborators, from the receipt of license fees and royalties from sales of licensed products. We conduct our research and development programs through a combination of internal and collaborative programs. We rely on arrangements with universities, certain collaborators, contract development and manufacturing organizations (“CDMOs”), clinical research organizations ("CROs") and clinical research sites for a significant portion of our product development efforts.
We focus substantially all of our research and development efforts in the development of simufilam. Research and development expenses for our investigational diagnostic product candidate, SavaDx, for which development was discontinued in mid-2025, represented less than 1% of total research and development expenses for the periods presented. The following table summarizes expenses by category for research and development efforts (in thousands):
| Three months ended |
||||||||
| March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Clinical trials |
$ | 102 | $ | 8,095 | ||||
| Pre-clinical projects |
959 | 715 | ||||||
| Chemical, Manufacturing and Controls costs (“CMC costs”) |
703 | 383 | ||||||
| Personnel related |
1,103 | 1,725 | ||||||
| Stock-based compensation |
1,333 | 2,372 | ||||||
| Other |
344 | 376 | ||||||
| $ | 4,544 | $ | 13,666 | |||||
Clinical trial costs include the costs of our CRO. CMC costs include costs related to our contract development and manufacturing organizations. Research and development expenses include compensation, contractor fees and supplies as well as allocated common costs such as facilities.
Estimating the dates of completion of clinical development, and the costs to complete development, of our product candidates would be highly speculative and subjective. Pharmaceutical products take a significant amount of time to research, develop and commercialize. The clinical study portion of the development of a new drug alone usually spans several years. In the near term, we expect research and development expense for the TSC-related epilepsy program to be significantly lower compared to those for the Alzheimer's disease program. We expect to reassess our future research and development plans based on our review of data we receive from our current research and development activities. The cost and pace of our future research and development activities are linked and subject to change.
Critical Accounting Estimates
This discussion and analysis of our financial condition and results of operations is based on our condensed consolidated financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these condensed consolidated financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the condensed consolidated financial statements, as well as the reported expenses and net loss incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
There have been no material changes to our critical accounting estimates during the three months ended March 31, 2026 from those described in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included in our Annual Report on Form 10-K for the year ended December 31, 2025, as filed with the SEC on March 12, 2026.
Results of Operations – Three Months Ended March 31, 2026 and 2025
Research and Development Expense
Research and development expenses consist primarily of costs of drug development work associated with our product candidates, including:
| ● |
clinical trials, |
|
| ● |
pre-clinical testing, |
|
| ● |
clinical supplies and related formulation and design costs, and |
|
| ● |
compensation and other personnel-related expenses. |
Research and development expenses were $4.5 million and $13.7 million during the three months ended March 31, 2026 and 2025, respectively. This 67% decrease was due primarily to the phase out of the Alzheimer's disease development program, which was completed in the second quarter of 2025.
In the near term, we expect research and development expense for the TSC-related epilepsy program to be significantly lower compared to those for the Alzheimer's disease program.
General and Administrative Expense
General and administrative expenses consist of personnel costs, allocated expenses and other expenses for outside professional services, including legal, human resources, audit and accounting services. Personnel costs consist of salaries, bonus, benefits and stock-based compensation. Allocated expenses consist primarily of facility costs for our Company owned office complex in Austin, Texas. Depreciation and amortization for office space leased but not occupied by the Company is included in general and administrative expense. Depreciation and amortization for office space occupied by the Company is allocated between general and administrative expense and research and development expense. We also incur expenses associated with operating as a public company, including additional legal fees, expenses related to compliance with the rules and regulations of the SEC and Nasdaq, additional insurance and audit expenses, investor relations activities, public company compliance expenses and other administrative expenses and professional services.
General and administrative expenses were $6.6 million and $10.9 million during the three months ended March 31, 2026 and 2025, respectively. The 39% decrease was due primarily to legal loss contingencies of $3.0 million recorded in the first quarter of 2025 not being repeated in 2026.
We expect general and administrative expense will decrease in future periods. However, we expect general and administrative expense to remain high compared to historic levels due to professional fees, legal expense and potential settlements related to ongoing litigation.
Interest Income
Interest income was $0.8 million and $1.3 million during the three months ended March 31, 2026 and 2025, respectively. The decrease in interest income was due primarily to lower interest rates and cash balances in 2026 compared to 2025.
We expect interest income to decrease in future quarters as we utilize cash in our operations and realize the impact of a lower interest rate environment.
Other income (loss), net
We record the activities related to leasing office space to third parties in buildings we own as other income (loss), net, as leasing is not core to the Company’s operations. Other income (loss), net, was $48,000 and $(82,000) during the three months ended March 31, 2026 and 2025, respectively.
We recorded other income in the three months ended March 31, 2026 due to improved occupancy rates in 2026 compared to the prior year period. We expect to record modest net income on leasing activities in 2026 due to improved occupancy.
Depreciation and amortization for the office complex is included in general and administrative and research and development expense, and thus not reflected in other income, net.
Liquidity and Capital Resources
Since inception, we have financed our operations primarily through public and private stock offerings, payments received under collaboration agreements and interest earned on our cash and cash equivalents balances. We intend to continue to use our capital resources to fund research and development activities, capital expenditures, working capital requirements and other general corporate purposes. As of March 31, 2026, cash and cash equivalents were $86.6 million.
At the Market (ATM) Common Stock Issuance
On November 12, 2025, we entered into an at-the-market offering program (ATM) to sell, from time to time, shares of our common stock having an aggregate offering price of up to $50 million in common stock in transactions pursuant to a shelf registration statement that was declared effective by the U.S. Securities and Exchange Commission (the SEC) on December 5, 2025. We are obligated to pay a commission of up to 3.0% of the gross proceeds from the sale of shares of common stock in the offering. We are not obligated to sell any shares in the offering.
There were no common stock sales under the ATM during the three months ended March 31, 2026 or the year ended December 31, 2025.
2020 Cash Incentive Bonus Plan Obligations
Information with respect to the 2020 Cash Incentive Bonus Plan is included in Note 10 of “Notes to Condensed Consolidated Financial Statements” included in Part I, Item 1 of this Quarterly Report on Form 10-Q.
No cash payments were authorized or made to participants under the CIB Plan as of March 31, 2026, or through the filing date of this Quarterly Report on Form 10-Q.
Use of Cash
The following table sets forth a summary of the primary sources and uses of cash for each of the periods presented below (in thousands):
| Three months ended March 31, |
||||||||
| 2026 |
2025 |
|||||||
| Net cash used in operating activities |
$ | (8,878 | ) | $ | (11,336 | ) | ||
| Net cash used in investing activities |
— | — | ||||||
| Net cash provided by (used in) financing activities |
(51 | ) | 90 | |||||
| Net decrease in cash and cash equivalents |
$ | (8,929 | ) | $ | (11,246 | ) | ||
Net cash used in operating activities was $8.9 million for the three months ended March 31, 2026, resulting primarily from a net loss of $10.3 million as well as decreases in accounts payable and accrued expenses of $2.5 million and accrued compensation and benefits of $1.4 million. These factors were partially offset by a decrease in prepaid and other current assets of $0.9 million. There was also a non-cash adjustment for stock-based compensation expense of $4.1 million.
Net cash used in operating activities was $11.3 million for the three months ended March 31, 2025, resulting primarily from a net loss of $23.4 million. Impacts from this net loss were partially offset by an increase in accrued development expense of $1.6 million, a decrease in prepaid and other current assets of $5.1 million, and non-cash adjustment for stock-based compensation expense of $5.2 million.
There was no net cash used in investing activities during the three months ended March 31, 2026 and 2025.
Net cash used in financing activities during the three months ended March 31, 2026 was $51,000 for costs related to the ATM. There were no sales of common stock under the ATM during the three months ended March 31, 2026.
Net cash provided by financing activities during the three months ended March 31, 2025 was $90,000 of proceeds from the exercise of stock options.
Property and Leases
We own an office complex in Austin, Texas, a portion of which serves as our corporate headquarters. Maintenance, physical facilities, leasing, property management and other key responsibilities related to property ownership are outsourced to professional real-estate managers. The office complex measures approximately 90,000 rentable square feet. At March 31, 2026, we occupied approximately 25% of the property with the remainder either leased or available for lease to third parties. We expect to record modest net income on leasing activities in 2026 due to improved occupancy compared to the prior year.
Other Commitments
We have an accumulated deficit of $506.4 million as of March 31, 2026. We expect our cash requirements to be significant in the future. The amount and timing of our future cash requirements will depend on regulatory and market acceptance of our drug candidates, the resources we devote to researching and developing, formulating, manufacturing, commercializing and supporting our products and other corporate needs. We believe that our current cash and cash equivalents will be sufficient to fund our operations for at least the next 12 months. We may seek additional future funding through public or private financing in the future, if such funding is available and on terms acceptable to us. However, there are no assurances that additional financing will be available on favorable terms, or at all.
Item 3. Quantitative and Qualitative Disclosures About Market Risk
As a smaller reporting company, per Item 305(e) of Regulation S-K, we are not required to provide the information called for by this Item 3.
Item 4. Controls and Procedures
Evaluation of disclosure controls and procedures. Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Quarterly Report on Form 10-Q. Based on this evaluation, our Chief Executive Officer (as Principal Executive Officer) and our Chief Financial Officer (as Principal Financial Officer) have concluded that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosures.
Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of March 31, 2026, our Chief Executive Officer and Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Changes in internal control over financial reporting. There has been no change in our internal control over financial reporting (as defined in Rule 13a-15(f) under the Exchange Act) identified during the three months ended March 31, 2026 that has materially affected, or is reasonable likely to materially affect, our internal control over financial reporting.
PART II – OTHER INFORMATION
We are, and from time to time, we may become involved in litigation or other legal proceedings and claims, including U.S. government inquiries, investigations and Citizen Petitions submitted to FDA. In addition, we have received and from time to time may receive inquiries from government authorities relating to matters arising from the ordinary course of business. The outcome of these proceedings is inherently uncertain. Regardless of outcome, legal proceedings can have an adverse impact on us because of defense and settlement costs, diversion of management resources, and other factors. At this time, no assessment can be made as to their likely outcome or whether the outcome will be material to us except as disclosed in this Quarterly Report on Form 10-Q in Note 10 to our condensed consolidated financial statements entitled, “Contingencies.” We believe that our total provisions for legal matters are adequate based upon currently available information. Additional information regarding our legal proceedings is included in the “Contingencies” note.
Please refer to “Risk Factors” in Part I, Item 1A of our 2025 Annual Report on Form 10‑K for additional information on our current risks. There have been no material changes in our risk factors from those disclosed in our Annual Report on Form 10-K. The risks and uncertainties described in our 2025 Annual Report on Form 10‑K are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also materially adversely affect our business, financial condition or results of operations.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds
None.
Item 3. Defaults Upon Senior Securities
None.
Item 4. Mine Safety Disclosures
Not applicable.
During the quarter ended March 31, 2026, of our directors or officers (as defined in Rule 16a-1(f) of the Exchange Act) informed us of the adoption or termination of a “Rule 10b5-1 trading arrangement” or “non-Rule 10b5-1 trading arrangement,” as those terms are defined in Regulation S-K, Item 408.
The following exhibits have been filed with this report:
| Incorporated by |
|||||||||
| Reference |
|||||||||
| Exhibit |
Description |
Filing |
Exhibit |
Filed |
|||||
| No. |
Form |
Date |
No. |
Herewith |
|||||
| 3.1 |
10-Q |
7/29/2005 |
3.1 |
||||||
| 3.2 |
Certificate of Amendment of Restated Certificate of Incorporation. |
8-K |
5/8/2017 |
3.1 |
|||||
| 3.3 |
Certificate of Amendment of Restated Certificate of Incorporation. |
10-K |
3/29/2019 |
3.3 |
|||||
| 3.4 | Certificate of Amendment of Restated Certificate of Incorporation. | 8-K | 3/10/2026 | 3.4 | |||||
| 3.5 |
8-K | 3/10/2026 | 3.5 |
||||||
| 31.1 |
X |
||||||||
| 31.2 |
X |
||||||||
| 32.1+ |
X |
||||||||
| 101.INS |
Inline XBRL Instance Document - (the instant document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document). |
X |
|||||||
| 101.SCH |
Inline XBRL Taxonomy Extension Schema Document. |
X |
|||||||
| 101.CAL |
Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
X |
|||||||
| 101.DEF |
Inline XBRL Taxonomy Extension Definition Linkbase Document. |
X |
|||||||
| 101.LAB |
Inline XBRL Taxonomy Extension Labels Linkbase Document. |
X |
|||||||
| 101.PRE |
Inline XBRL Taxonomy Extension Presentation Linkbase Document. |
X |
|||||||
| 104 |
Cover Page Interactive Data File –(formatted as Inline XBRL and contained in Exhibit 101). |
X |
|||||||
+The certification furnished in Exhibit 32.1 hereto is deemed to accompany this Quarterly Report on Form 10-Q and will not be deemed “filed” for purposes of Section 18 of the Exchange Act of 1934. Such certification will not be deemed to be incorporated by reference into any filings under the Securities Act of 1933, as amended, or the Exchange Act, except to the extent that the Company specifically incorporates it by reference.
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| Filana Therapeutics, Inc. |
|
| (Registrant) |
|
| /s/ RICHARD J. BARRY |
|
| Richard J. Barry, |
|
| President and Chief Executive Officer |
|
| Date: May 7, 2026 |
(Principal Executive Officer) |
| /s/ ERIC J. SCHOEN |
|
| Eric J. Schoen, |
|
| Chief Financial Officer |
|
| Date: May 7, 2026 | (Principal Financial and Accounting Officer) |