Please wait
false
FY
0001326706
http://fasb.org/us-gaap/2025#LeaseholdImprovementsMember
http://fasb.org/us-gaap/2025#OperatingExpenses
http://fasb.org/us-gaap/2025#OperatingExpenses
http://xbrl.sec.gov/country/2025#IL
http://fasb.org/us-gaap/2025#OperatingExpenses
0001326706
2025-01-01
2025-12-31
0001326706
2025-06-30
0001326706
2026-04-15
0001326706
2025-12-31
0001326706
2024-12-31
0001326706
us-gaap:SeriesCPreferredStockMember
2025-12-31
0001326706
us-gaap:SeriesCPreferredStockMember
2024-12-31
0001326706
us-gaap:SeriesHPreferredStockMember
2025-12-31
0001326706
us-gaap:SeriesHPreferredStockMember
2024-12-31
0001326706
FEED:SeriesXPreferredStockMember
2025-12-31
0001326706
FEED:SeriesXPreferredStockMember
2024-12-31
0001326706
us-gaap:SeriesGPreferredStockMember
2025-12-31
0001326706
us-gaap:SeriesGPreferredStockMember
2024-12-31
0001326706
2024-01-01
2024-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesGPreferredStockMember
2023-12-31
0001326706
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2023-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesHPreferredStockMember
2023-12-31
0001326706
us-gaap:CommonStockMember
2023-12-31
0001326706
us-gaap:AdditionalPaidInCapitalMember
2023-12-31
0001326706
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2023-12-31
0001326706
us-gaap:RetainedEarningsMember
2023-12-31
0001326706
2023-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesGPreferredStockMember
2024-12-31
0001326706
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2024-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesHPreferredStockMember
2024-12-31
0001326706
us-gaap:CommonStockMember
2024-12-31
0001326706
us-gaap:AdditionalPaidInCapitalMember
2024-12-31
0001326706
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-12-31
0001326706
us-gaap:RetainedEarningsMember
2024-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesGPreferredStockMember
2024-01-01
2024-12-31
0001326706
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2024-01-01
2024-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesHPreferredStockMember
2024-01-01
2024-12-31
0001326706
us-gaap:CommonStockMember
2024-01-01
2024-12-31
0001326706
us-gaap:AdditionalPaidInCapitalMember
2024-01-01
2024-12-31
0001326706
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2024-01-01
2024-12-31
0001326706
us-gaap:RetainedEarningsMember
2024-01-01
2024-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesGPreferredStockMember
2025-01-01
2025-12-31
0001326706
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2025-01-01
2025-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesHPreferredStockMember
2025-01-01
2025-12-31
0001326706
us-gaap:CommonStockMember
2025-01-01
2025-12-31
0001326706
us-gaap:AdditionalPaidInCapitalMember
2025-01-01
2025-12-31
0001326706
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2025-01-01
2025-12-31
0001326706
us-gaap:RetainedEarningsMember
2025-01-01
2025-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesGPreferredStockMember
2025-12-31
0001326706
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2025-12-31
0001326706
us-gaap:PreferredStockMember
us-gaap:SeriesHPreferredStockMember
2025-12-31
0001326706
us-gaap:CommonStockMember
2025-12-31
0001326706
us-gaap:AdditionalPaidInCapitalMember
2025-12-31
0001326706
us-gaap:AccumulatedOtherComprehensiveIncomeMember
2025-12-31
0001326706
us-gaap:RetainedEarningsMember
2025-12-31
0001326706
srt:MaximumMember
2025-12-31
0001326706
srt:MinimumMember
2025-01-01
2025-12-31
0001326706
srt:MaximumMember
2025-01-01
2025-12-31
0001326706
srt:ChiefFinancialOfficerMember
2025-01-01
2025-12-31
0001326706
FEED:PurchaseAgreementMember
2025-01-01
2025-12-31
0001326706
FEED:PurchaseAgreementMember
2025-10-01
2025-12-31
0001326706
FEED:PurchaseAgreementMember
2025-12-31
0001326706
FEED:EmployeeOptionsMember
2025-01-01
2025-12-31
0001326706
us-gaap:TrademarksAndTradeNamesMember
2025-12-31
0001326706
us-gaap:TechnologyBasedIntangibleAssetsMember
2025-12-31
0001326706
us-gaap:CustomerListsMember
2025-12-31
0001326706
us-gaap:ComputerEquipmentMember
2025-12-31
0001326706
us-gaap:OfficeEquipmentMember
srt:MinimumMember
2025-12-31
0001326706
us-gaap:OfficeEquipmentMember
srt:MaximumMember
2025-12-31
0001326706
FEED:MergerAgreementMember
us-gaap:CommonStockMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
us-gaap:CommonStockMember
srt:MaximumMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
FEED:PreFundedWarrantsMember
srt:MaximumMember
2025-02-14
0001326706
FEED:MergerAgreementMember
FEED:PreFundedWarrantsMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
FEED:SeriesXPreferredStockMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
us-gaap:PreferredStockMember
FEED:SeriesXPreferredStockMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
2025-02-14
2025-02-14
0001326706
FEED:MergerAgreementMember
2025-01-01
2025-12-31
0001326706
srt:MinimumMember
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
FEED:CommonStockAndPreFundedWarrantsMember
2025-01-01
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
FEED:SeriesXPreferredStockMember
2025-01-01
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
2025-01-01
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
us-gaap:TrademarksAndTradeNamesMember
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
FEED:ProprietaryTechnologyMember
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
us-gaap:CustomerRelationshipsMember
2025-12-31
0001326706
FEED:ENvueMedicalHoldingsCorpMember
2024-01-01
2024-12-31
0001326706
us-gaap:TrademarksAndTradeNamesMember
2025-12-31
0001326706
us-gaap:TrademarksAndTradeNamesMember
2025-01-01
2025-12-31
0001326706
FEED:ProprietaryTechnologyMember
2025-12-31
0001326706
FEED:ProprietaryTechnologyMember
2025-01-01
2025-12-31
0001326706
us-gaap:CustomerRelationshipsMember
2025-12-31
0001326706
us-gaap:CustomerRelationshipsMember
2025-01-01
2025-12-31
0001326706
2025-03-13
2025-03-13
0001326706
2025-08-11
2025-08-11
0001326706
2025-03-13
0001326706
2025-08-11
0001326706
us-gaap:SeriesGPreferredStockMember
2025-05-15
2025-05-15
0001326706
us-gaap:SeriesGPreferredStockMember
2025-05-15
0001326706
2025-05-15
2025-05-15
0001326706
us-gaap:SeriesGPreferredStockMember
2025-05-16
2025-07-01
0001326706
us-gaap:SeriesGPreferredStockMember
2025-07-01
0001326706
FEED:SeriesXPreferredStockMember
2025-02-14
0001326706
FEED:JulyPurchaseAgreementMember
FEED:SeriesXPreferredStockMember
2025-07-22
0001326706
FEED:JulyPurchaseAgreementMember
FEED:SeriesXPreferredStockMember
FEED:JulyPrivatePlacementMember
2025-07-22
2025-07-22
0001326706
FEED:SeriesXPreferredStockMember
FEED:SeriesXAmendmentAgreementMember
2024-10-01
2024-12-31
0001326706
srt:MinimumMember
FEED:SeriesXPreferredStockMember
FEED:SeriesXAmendmentAgreementMember
2025-05-12
0001326706
srt:MaximumMember
FEED:SeriesXPreferredStockMember
FEED:SeriesXAmendmentAgreementMember
2025-05-12
0001326706
FEED:SeriesXPreferredStockMember
FEED:SeriesXAmendmentAgreementMember
2025-05-12
2025-05-12
0001326706
us-gaap:SeriesHPreferredStockMember
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
0001326706
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
0001326706
us-gaap:SeriesHPreferredStockMember
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
2025-07-18
0001326706
us-gaap:WarrantMember
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
0001326706
FEED:JulyTwoThousandTwentyFivePrivatePlacementMember
2025-07-22
2025-07-22
0001326706
FEED:JulyTwoThousandTwentyFivePrivatePlacementMember
us-gaap:SeriesHPreferredStockMember
2025-07-22
2025-07-22
0001326706
FEED:JulyTwoThousandTwentyFivePrivatePlacementMember
us-gaap:SeriesHPreferredStockMember
2025-12-31
0001326706
FEED:SeriesHAmendmentAgreementMember
2026-01-30
2026-01-30
0001326706
us-gaap:SeriesHPreferredStockMember
2025-07-22
2025-07-22
0001326706
us-gaap:SeriesHPreferredStockMember
2025-07-22
0001326706
us-gaap:SeriesHPreferredStockMember
2025-01-01
2025-12-31
0001326706
FEED:SecuritiesPurchaseAgreementMember
us-gaap:CommonStockMember
2025-09-16
2025-09-16
0001326706
FEED:SecuritiesPurchaseAgreementMember
FEED:PreFundedWarrantsMember
2025-09-16
2025-09-16
0001326706
FEED:SecuritiesPurchaseAgreementMember
us-gaap:CommonStockMember
2025-09-16
0001326706
FEED:SecuritiesPurchaseAgreementMember
FEED:PrefundedWarrantMember
2025-09-16
0001326706
FEED:SecuritiesPurchaseAgreementMember
2025-09-16
2025-09-16
0001326706
us-gaap:StockOptionMember
FEED:TwoThousandAndTwentyFourLongTermIncentivePlanMember
2024-12-31
0001326706
FEED:TwoThousandAndTwentyFourLongTermIncentivePlanMember
us-gaap:StockOptionMember
2025-03-14
0001326706
FEED:TwoThousandAndTwentyFourLongTermIncentivePlanMember
us-gaap:StockOptionMember
2025-01-01
2025-12-31
0001326706
us-gaap:StockOptionMember
2025-01-01
2025-12-31
0001326706
us-gaap:StockOptionMember
2024-01-01
2024-12-31
0001326706
us-gaap:CommonStockMember
2023-08-30
0001326706
FEED:SeriesA1WarrantsMember
2023-08-30
0001326706
FEED:SeriesA2WarrantsMember
2023-08-30
0001326706
2023-08-30
0001326706
us-gaap:CommonStockMember
FEED:SecuritiesExchangeAgreementMember
2025-01-07
2025-01-07
0001326706
FEED:PrefundedWarrantMember
2025-01-07
0001326706
FEED:PreFundedWarrantsMember
FEED:SecuritiesExchangeAgreementMember
2025-01-07
0001326706
FEED:SecuritiesExchangeAgreementMember
FEED:SeriesA1WarrantsMember
2025-01-07
0001326706
FEED:SecuritiesExchangeAgreementMember
FEED:SeriesA1WarrantsMember
2025-01-07
2025-01-07
0001326706
FEED:PrefundedWarrantMember
2025-02-24
0001326706
2025-01-07
2025-01-07
0001326706
us-gaap:WarrantMember
2025-01-07
2025-01-07
0001326706
us-gaap:StockOptionMember
2024-12-31
0001326706
us-gaap:StockOptionMember
2025-01-01
2025-03-31
0001326706
us-gaap:StockOptionMember
2025-10-01
2025-12-31
0001326706
us-gaap:StockOptionMember
2025-12-31
0001326706
2023-01-01
2023-12-31
0001326706
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
2025-01-01
2025-12-31
0001326706
us-gaap:MeasurementInputExpectedDividendRateMember
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
2025-12-31
0001326706
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
us-gaap:MeasurementInputExpectedTermMember
2025-12-31
0001326706
us-gaap:MeasurementInputPriceVolatilityMember
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
2025-12-31
0001326706
us-gaap:MeasurementInputRiskFreeInterestRateMember
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
2025-12-31
0001326706
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-01-01
2025-12-31
0001326706
us-gaap:MeasurementInputSharePriceMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
us-gaap:MeasurementInputExercisePriceMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
us-gaap:MeasurementInputExpectedDividendRateMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
FEED:MayTwoThousandTwentyFiveWarrantsAndRepresentativesWarrantsMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
us-gaap:MeasurementInputPriceVolatilityMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
us-gaap:MeasurementInputRiskFreeInterestRateMember
FEED:MayTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
FEED:JulyTwoThousandTwentyFiveWarrantsMember
2025-01-01
2025-12-31
0001326706
us-gaap:MeasurementInputExpectedDividendRateMember
FEED:JulyTwoThousandTwentyFiveWarrantsMember
2025-12-31
0001326706
FEED:JulyTwoThousandTwentyFiveWarrantsMember
us-gaap:MeasurementInputExpectedTermMember
2025-12-31
0001326706
us-gaap:MeasurementInputPriceVolatilityMember
FEED:JulyTwoThousandTwentyFiveWarrantsMember
2025-12-31
0001326706
us-gaap:MeasurementInputRiskFreeInterestRateMember
FEED:JulyTwoThousandTwentyFiveWarrantsMember
2025-12-31
0001326706
FEED:JulyTwoThousandTwentyFiveWarrantsOneMember
2025-01-01
2025-12-31
0001326706
us-gaap:MeasurementInputExpectedDividendRateMember
FEED:JulyTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
FEED:JulyTwoThousandTwentyFiveWarrantsOneMember
us-gaap:MeasurementInputExpectedTermMember
2025-12-31
0001326706
us-gaap:MeasurementInputPriceVolatilityMember
FEED:JulyTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
us-gaap:MeasurementInputRiskFreeInterestRateMember
FEED:JulyTwoThousandTwentyFiveWarrantsOneMember
2025-12-31
0001326706
FEED:OthersIncomeMember
2025-01-01
2025-12-31
0001326706
us-gaap:WarrantMember
2024-12-31
0001326706
us-gaap:WarrantMember
2025-01-01
2025-12-31
0001326706
us-gaap:WarrantMember
2025-12-31
0001326706
2025-02-13
0001326706
FEED:ENvueMedicalHoldingsCorpMember
2025-03-26
0001326706
srt:MinimumMember
2025-03-26
0001326706
FEED:ENvueConsolidatedSecuredNoteMember
2025-01-17
0001326706
FEED:ENvueConsolidatedSecuredNoteMember
FEED:ENvueMedicalHoldingsCorpMember
2025-12-31
2025-12-31
0001326706
FEED:AprilTwoThousandTwentyFivePromissoryNoteMember
2025-04-11
0001326706
FEED:AprilTwoThousandTwentyFivePromissoryNoteMember
2025-04-11
2025-04-11
0001326706
FEED:StockOptionsEmployeeAndNonEmployeeMember
2025-01-01
2025-12-31
0001326706
FEED:StockOptionsEmployeeAndNonEmployeeMember
2024-01-01
2024-12-31
0001326706
us-gaap:WarrantMember
2025-01-01
2025-12-31
0001326706
us-gaap:WarrantMember
2024-01-01
2024-12-31
0001326706
FEED:NanoVibronixMember
2025-12-31
0001326706
FEED:EnvueMember
2025-12-31
0001326706
us-gaap:RelatedPartyMember
2025-12-31
0001326706
us-gaap:CorporateMember
2025-12-31
0001326706
FEED:NanoVibronixMember
2024-12-31
0001326706
FEED:EnvueMember
2024-12-31
0001326706
us-gaap:CorporateMember
2024-12-31
0001326706
us-gaap:RelatedPartyMember
2024-12-31
0001326706
FEED:NanoVibronixMember
2025-01-01
2025-12-31
0001326706
FEED:EnvueMember
2025-01-01
2025-12-31
0001326706
us-gaap:CorporateMember
2025-01-01
2025-12-31
0001326706
us-gaap:RelatedPartyMember
2025-01-01
2025-12-31
0001326706
FEED:NanoVibronixMember
2024-01-01
2024-12-31
0001326706
FEED:EnvueMember
2024-01-01
2024-12-31
0001326706
us-gaap:CorporateMember
2024-01-01
2024-12-31
0001326706
us-gaap:RelatedPartyMember
2024-01-01
2024-12-31
0001326706
country:US
2025-01-01
2025-12-31
0001326706
country:US
2024-01-01
2024-12-31
0001326706
srt:EuropeMember
2025-01-01
2025-12-31
0001326706
srt:EuropeMember
2024-01-01
2024-12-31
0001326706
FEED:AustraliaNewZealandMember
2025-01-01
2025-12-31
0001326706
FEED:AustraliaNewZealandMember
2024-01-01
2024-12-31
0001326706
FEED:OtherCountryMember
2025-01-01
2025-12-31
0001326706
FEED:OtherCountryMember
2024-01-01
2024-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerAMember
2025-01-01
2025-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerAMember
2024-01-01
2024-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerBMember
2025-01-01
2025-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerBMember
2024-01-01
2024-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerCMember
2025-01-01
2025-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomerCMember
2024-01-01
2024-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomersMember
2025-01-01
2025-12-31
0001326706
us-gaap:SalesRevenueNetMember
us-gaap:CustomerConcentrationRiskMember
FEED:CustomersMember
2024-01-01
2024-12-31
0001326706
us-gaap:FairValueInputsLevel3Member
2024-12-31
0001326706
us-gaap:FairValueInputsLevel3Member
2025-01-01
2025-12-31
0001326706
us-gaap:FairValueInputsLevel3Member
2025-12-31
0001326706
us-gaap:FairValueInputsLevel1Member
2025-12-31
0001326706
us-gaap:FairValueInputsLevel2Member
2025-12-31
0001326706
2021-02-25
2021-02-26
0001326706
2022-03-14
2022-03-15
0001326706
FEED:ArbitratorMember
2025-01-01
2025-12-31
0001326706
FEED:PiersonFerdinandMember
2025-01-01
2025-12-31
0001326706
FEED:PiersonFerdinandMember
2024-01-01
2024-12-31
0001326706
FEED:SeriesXPreferredStockMember
2025-01-01
2025-12-31
0001326706
FEED:SeriesHConvertiblePreferredStockMember
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
0001326706
us-gaap:SeriesHPreferredStockMember
srt:MaximumMember
FEED:JulyTwoThousandTwentyFivePurchaseAgreementMember
2025-07-18
2025-07-18
0001326706
country:IL
2025-01-01
2025-12-31
0001326706
country:IL
2024-01-01
2024-12-31
0001326706
us-gaap:DomesticCountryMember
2025-12-31
0001326706
us-gaap:StateAndLocalJurisdictionMember
2025-12-31
0001326706
us-gaap:DomesticCountryMember
2025-01-01
2025-12-31
0001326706
us-gaap:SubsequentEventMember
FEED:EmploymentAgreementMember
FEED:ENvueMedicalIsraelLtdMember
2026-02-02
0001326706
us-gaap:SubsequentEventMember
FEED:ChairmanAgreementMember
2026-02-02
0001326706
us-gaap:SubsequentEventMember
2026-03-25
iso4217:USD
xbrli:shares
iso4217:USD
xbrli:shares
xbrli:pure
FEED:Integer
UNITED
STATES
SECURITIES
AND EXCHANGE COMMISSION
Washington,
D.C. 20549
FORM
10-K
☒
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For
the fiscal year ended December 31, 2025
OR
☐
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For
the transition period from to
Commission
File Number: 001-36445
ENvue
Medical, Inc.
(Exact
name of registrant as specified in its charter)
| Delaware |
|
01-0801232 |
(State
or other jurisdiction of
incorporation
or organization) |
|
(I.R.S.
Employer
Identification
Number) |
| |
|
|
969
Pruitt Ave
Tyler, Texas |
|
77569 |
| (Address
of principal executive office) |
|
(Zip
Code) |
Registrant’s
telephone number, including area code: (914) 233-3004
Securities
registered pursuant to Section 12(b) of the Act:
| Title
of each class |
|
Trading
Symbol |
|
Name
of each exchange on which registered |
| Common
stock, par value $0.001 per share |
|
FEED |
|
NASDAQ
Capital Market |
Securities
registered pursuant to Section 12(g) of the Act: None
Indicate
by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate
by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate
by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange
Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2)
has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate
by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule
405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant
was required to submit such files). Yes ☒ No ☐
Indicate
by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting
company or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,”
“smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| Large
accelerated filer |
☐ |
Accelerated
filer |
☐ |
| Non-accelerated
filer |
☒ |
Smaller
reporting company |
☒ |
| |
|
Emerging
growth company |
☐ |
If
an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying
with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate
by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness
of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C 7262(b)) by the registered
public accounting firm that prepared or issued its audit report. ☐
If
securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant
included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate
by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation
received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate
by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The
aggregate market value of voting stock held by non-affiliates as of June 30, 2025, the last business day of the registrant’s most
recently completed second quarter and based on the closing price of the registrant’s common stock as reported on the Nasdaq Capital
Market, was approximately $7.3 Million.
The
number of shares outstanding of the registrant’s common stock as of April 15, 2026, was 3,700,908 shares.
DOCUMENTS
INCORPORATED BY REFERENCE
None.
TABLE
OF CONTENTS
EXPLANATORY
NOTE
On
February 14, 2025, subsequent to the end of the fiscal year ended December 31, 2024, the fiscal year to which this Annual Report on Form
10-K relates and as further described herein, pursuant to the terms of that certain Agreement and Plan of Merger, dated as of February
14, 2025 (the “Merger Agreement”), by and among ENvue Medical, Inc. (formerly NanoVibronix, Inc.) (the “Company”)
NVEH Merger Sub I, Inc., a Delaware corporation and a wholly-owned subsidiary of the Company (“First Merger Sub I”), NVEH
Merger Sub II, LLC, a Delaware limited liability company and a wholly-owned subsidiary of the Company (“Second Merger Sub”),
and ENvue Medical Holdings LLC (“Predecessor ENvue”), the Company and Predecessor ENvue effected (i) a merger of First Merger
Sub with and into Predecessor ENvue, with the First Merger Sub ceasing to exist and Predecessor ENvue becoming a wholly-owned subsidiary
of the Company and (ii) the merger of Predecessor ENvue with and into Second Merger Sub (the “Second Merger” and, together
with the First Merger, the “Merger”), with Second Merger Sub being the surviving entity of the Second Merger (“Surviving
Entity”). At the effective time of the Second Merger, the certificate of formation of the Surviving Entity was amended and restated
to, among other things, to change the name of the Surviving Entity to “ENvue Medical Holdings LLC.”
Cautionary
Note Regarding Forward-Looking Statements
This
Annual Report on Form 10-K contains “forward-looking statements,” which include information relating to future events, future
financial performance, financial projections, strategies, expectations, competitive environment and regulation. Words such as “may,”
“should,” “could,” “would,” “predicts,” “potential,” “continue,”
“expects,” “anticipates,” “future,” “intends,” “plans,” “believes,”
“estimates,” and similar expressions, as well as statements in future tense, are intended to identify forward-looking statements.
Forward-looking statements should not be read as a guarantee of future performance or results and may not be accurate indications of
when such performance or results will actually be achieved. Forward-looking statements are based on information we have when those statements
are made or our management’s good faith belief as of that time with respect to future events, and are subject to a number of risks,
and uncertainties and assumptions that could cause actual performance or results to differ materially from those expressed in or suggested
by the forward-looking statements. These risks are more fully described in the “Risk Factors” section of this Annual Report
on Form 10-K. Important factors that could cause such differences include, but are not limited to:
| |
● |
Our
history of losses and expectation of continued losses. |
| |
|
|
| |
● |
Global
economic and political instability and conflicts, such as the conflict between Russia and Ukraine, could adversely affect our business,
financial condition or results of operations. |
| |
|
|
| |
● |
Increasing
inflation could adversely affect our business, financial condition, results of operations or cash flows. |
| |
|
|
| |
● |
Risks
related to ENvue’s financial condition, business and operations, as well as legal, regulatory and compliance matters |
| |
|
|
| |
● |
Our
ability to raise funding for, and the timing of, clinical studies and eventual U.S. Food and Drug Administration (“FDA”)
approval of our product candidates. |
| |
|
|
| |
● |
Regulatory
actions that could adversely affect the price of or demand for our approved products. |
| |
|
|
| |
● |
Market
acceptance of existing and new products. |
| |
|
|
| |
● |
Favorable
or unfavorable decisions about our products from government regulators, insurance companies or other third-party payers (including
CMS). |
| |
|
|
| |
● |
Risks
of product liability acclaims and the availability of insurance.
|
| |
|
|
| |
● |
Our
ability to generate internal growth. |
| |
|
|
| |
● |
Risks
related to computer system failures and cyber-attacks. |
| |
|
|
| |
● |
Our
ability to obtain regulatory approval in foreign jurisdictions. |
| |
|
|
| |
● |
Uncertainty
regarding the success of our clinical trials for our products in development. |
| |
|
|
| |
● |
Risks
related to our operations in Israel, including political, economic and military instability. |
| |
|
|
| |
● |
The
price of our securities is volatile with limited trading volume. |
| |
|
|
| |
● |
Our
ability to regain and maintain compliance with the continued listing requirements of Nasdaq and the risk that our common stock will
be delisted if we cannot do so. |
| |
|
|
| |
● |
Our
ability to maintain effective internal control over financial reporting and to remedy identified material weaknesses. |
| |
|
|
| |
● |
We
are a “smaller reporting company” and have reduced disclosure obligations that may make our stock less attractive to
investors. |
| |
|
|
| |
● |
Our
intellectual property portfolio and our ability to protect our intellectual property rights. |
| |
|
|
| |
● |
Our
ability to recruit and retain qualified regulatory and research and development personnel. |
| |
● |
Unforeseen
changes in healthcare reimbursement for any of our approved products. |
| |
|
|
| |
● |
The
adoption of health policy changes and health care reform. |
| |
|
|
| |
● |
Lack
of financial resources to adequately support our operations. |
| |
|
|
| |
● |
Difficulties
in maintaining commercial scale manufacturing capacity and capability. |
| |
|
|
| |
● |
Changes
in our relationship with key collaborators. |
| |
|
|
| |
● |
Changes
in the market valuation or earnings of our competitors or companies viewed as similar to us. |
| |
|
|
| |
● |
Our
failure to comply with regulatory guidelines. |
| |
|
|
| |
● |
Uncertainty
in industry demand and patient wellness behavior. |
| |
|
|
| |
● |
General
economic conditions and market conditions in the medical device industry. |
| |
|
|
|
|
● |
Future
sales of large blocks of our common stock, which may adversely impact our stock price. |
| |
|
|
| |
● |
Depth
of the trading market in our common stock. |
The
foregoing does not represent an exhaustive list of matters that may be covered by the forward-looking statements contained herein or
risk factors that we are faced with that may cause our actual results to differ from those anticipated in our forward-looking statements.
Please see “Item 1A. Risk Factors” for additional risks which could adversely impact our business and financial performance.
Moreover, new risks regularly emerge, and it is not possible for us to predict or articulate all risks we face, nor can we assess the
impact of all risks on our business or the extent to which any risk, or combination of risks, may cause actual results to differ from
those contained in any forward-looking statements. All forward-looking statements included in this Annual Report on Form 10-K are based
on information available to us on the date hereof. Except to the extent required by applicable laws or rules, we undertake no obligation
to publicly update or revise any forward-looking statement, whether as a result of new information, future events or otherwise.
Unless
the context otherwise indicates or requires, the terms “we,” “our,” “us,”, “ENvue”,
“NanoVibronix,” and the “Company,” as used in this Annual Report on Form 10-K, refer to ENvue Medical, Inc.
and its subsidiaries as a combined entity, except where otherwise stated or where it is clear that the terms mean only ENvue
Medical, Inc. exclusive of its subsidiaries.
Trademarks
We
have proprietary rights to certain trademarks used in this Annual Report on Form 10-K that are important to our business, some of which
are registered under applicable intellectual property laws, including but not limited to UroShield™, PainShield™ MD, PainShield™
Plus, WoundShield™, UroShield®, PainShield®, PainShield Plus®, WoundShield®, UroShield®, NanoVibronix®,
ENvue Medical, ENsump, ENvue, ENgat, ENvue (wordmark and logo), and ENvue’s logo in key countries, including the U.S., Europe,
and China.
Solely
for convenience, trademarks and trade names referred to in this Annual Report appear without the “®” or “™”
symbols, but such references are not intended to indicate, in any way, that we will not assert, to the fullest extent possible under
applicable law, our rights or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or
display of other companies’ trademarks, trade names or service marks to imply a relationship with, or endorsement or sponsorship
of us by, any other companies. Each trademark, trade name or service mark of any other company appearing in this Annual Report on Form
10-K is the property of its respective holder.
PART
I
ITEM
1. BUSINESS
Overview
ENvue
Medical, Inc. (the “Company”), formerly known as NanoVibronix, Inc., was incorporated as a Delaware corporation in
October 2003. In December 2025, the Company changed its name from NanoVibronix, Inc. to ENvue Medical, Inc. Prior to such a name
change, on February 14, 2025, the Company consummated and completed its merger (the “Merger”) pursuant to the Agreement
and Plan of Merger, dated as of February 14, 2025 (the “Merger Agreement”), as further described herein. Following the
consummation of the Merger, the Company conducts its operations through two wholly-owned subsidiaries: (i) NanoVibronix Ltd.
(“Nano OpCo”), a private company incorporated under the laws of the State of Israel, which focuses on non-invasive,
biological response-activating medical devices targeting biofilm prevention and pain therapy, designed for home use without the need
for medical professional assistance; and (ii) ENvue Medical Holdings LLC, a Delaware limited liability company, which is a medical
device company engaged in the research, development, production, marketing, and sale of medical devices in the field of enteral
feeding, currently in the initial growth stage of commercialization. Further descriptions of each business division, their
respective products, and business models are set forth below.
The
Merger Agreement
On
February 14, 2025, pursuant to the terms of that certain Agreement and Plan of Merger (as amended, restated, amended and restated, supplemented
or modified from time to time, the “Merger Agreement”), dated as of February 14, 2025, by and among us, NVEH Merger Sub I,
Inc., a Delaware corporation and a wholly-owned subsidiary of the Company (“First Merger Sub”), NVEH Merger Sub II, LLC,
a Delaware limited liability company and a wholly-owned subsidiary of the Company (“Second Merger Sub”), and ENvue Medical
Holdings, Corp. (“Predecessor ENvue” or “ENvue”), the Company and Predecessor ENvue effected (i) a merger of
First Merger Sub with and into Predecessor ENvue, with the First Merger Sub ceasing to exist and Predecessor ENvue becoming a wholly-owned
subsidiary the Company (the “First Merger”, and effective time of such First Merger, the “First Effective Time”)
and (ii) the merger of Predecessor ENvue with and into Second Merger Sub (the “Second Merger” and, together with the First
Merger, the “Merger”), with Second Merger Sub being the surviving entity of the Second Merger (“Surviving Entity”).
At the effective time of the Second Merger, the certificate of formation of the Surviving Entity was amended and restated to, among other
things, to change the name of the Surviving Entity to “ENvue Medical Holdings LLC.” In connection with the Merger Agreement,
we issued (i) 3,318 shares of common stock (the “Merger Shares”), which such number of shares represented no more than 4.9%
(the “Exchange Cap”) of the outstanding shares of common stock as of immediately before the First Effective Time and (ii)
Pre-Funded Warrants to purchase up to 12,526 shares of our common stock (the “Merger Pre-Funded Warrants”) at an exercise
price of $0.001 per share, and (iii) 57,720 shares of Series X Non-Voting Convertible Preferred Stock (the “Series X Preferred
Stock”) in excess of the Exchange Cap to the holders of Predecessor ENvue in consideration for 100% of Predecessor ENvue.
2025
Reverse Stock Splits
On
March 12, 2025, the Company filed a Certificate of Amendment to its Certificate of Incorporation with the Secretary of State of Delaware
to effect a 1-for-11 reverse stock split of the shares of its common stock either issued and outstanding or held by the Company as treasury
stock, effective as of 4:05 p.m. (Delaware time) on March 13, 2025 (the “March 2025 Reverse Stock Split”).
On
August 8, 2025, the Company filed a Certificate of Amendment to its Certificate of Incorporation with the Secretary of State of Delaware
to effect a 1-for-10 reverse stock split of the shares of its common stock either issued and outstanding or held by the Company as treasury
stock, effective as of 4:05 p.m. (Delaware time) on August 11, 2025 (the “August 2025 Reverse Stock Split” and together with
the March 2025 Reverse Stock Split, the “Reverse Stock Splits”).
ENvue’s
Business
ENvue
is a medical device company engaged in the research, development, production, marketing, and sale of medical devices in the field of
enteral feeding and is in the growth stage of commercializing its products. Guided by its mission to be an innovation leader in the field
of enteral feeding, ENvue is focused on improving patient outcomes across the continuum of care, encompassing the development of advanced,
personalized navigation solutions, responding to the challenges of the ever-hanging healthcare environment, while continuously focusing
on the customer. The medical device marketed and sold by ENvue is the FDA 510(k)-cleared ENvue System, which assists in the insertion
of a feeding tube into the digestive system of patients requiring nutrition during hospitalization through intra-body navigation (the
“ENvue System”).
The
most common way to provide nutrition to patients during hospitalization is through a feeding tube inserted through the nose or mouth
into the stomach or small intestine (known as “enteral nutrition”). Around 43 million feeding tubes are inserted annually
worldwide (Enteral feeding devices, Global forecast to 2025; Market & Markets (“Markets & Markets”)). Between 2-5%
of these tubes are placed in the lungs leading to a 30% chance of a collapsed lung or possible fatality (Aguilar-Nascimento, Kudsk, JPEN
J Parenter Enteral Nutr 2007; Bourgault, Margo Halm Am J Crit Care. 2009). Furthermore, between 20-50% of hospital patients (Bellanti,
Francesco, et al. “Malnutrition in hospitalized old patients: screening and diagnosis, clinical outcomes, and management.”
Nutrients 14.4 (2022): 910.), including those in the intensive care unit (“ICU”), are malnourished, with malnutrition having
a significant impact on both clinical outcomes and healthcare systems.
Recognizing
the critical need for early feeding in small bowel and lower the risk of tube misplacement, ENvue applied its expertise in electromagnetic
navigation and enteral feeding to develop the ENvue System. The ENvue System, together with a dedicated feeding tube for the system,
positioning sensors, and other components developed by ENvue, is designed to efficiently and safely insert the feeding tube into the
patient’s digestive system for the purpose of providing nutrition. Furthermore, ENvue’s solution aims to provide faster nutrition
delivery to the patient, potentially improving their condition, and facilitating the insertion of the feeding tube into the small intestine,
which we believe has advantages over insertion into the stomach. ENvue believes that the ENvue System offers an efficient solution for
feeding tube insertion and has the ability to transform enteral feeding tube insertion.
In
February 2019, ENvue received 510(k) market clearance from U.S. Food and Drug Administration for the commercial marketing and sale
of the ENvue System in the United States for use in adults (aged 22 and over)1. During the first quarter of 2020, ENvue
began marketing and selling the ENvue System and its dedicated feeding tubes, and it is currently in the initial growth
commercialization phase of these products in the U.S.
1
According to FDA guidelines, an adult is defined as a person who is 22 years of age or older.
ENvue
Strategy
ENvue’s
strategy is focused on improving the safety, efficiency, and reliability of enteral feeding tube placement and management through the
development and commercialization of navigation-enabled medical devices and related disposable products. The Company is developing a
platform intended to assist clinicians in the real-time placement and verification of enteral feeding tubes, addressing longstanding
clinical challenges associated with traditional placement methods that may rely on blind insertion techniques and subsequent confirmation
procedures.
The
Company’s current commercial efforts are primarily focused on the United States acute care hospital market. ENvue is in the growth
commercialization phase of the ENvue System, a navigation platform designed to provide clinicians with real-time guidance during
feeding tube placement. The system is used in conjunction with proprietary disposable enteral feeding tubes designed to integrate with
the platform.
In
addition to feeding tubes, the Company offers enteral feeding accessories, including syringes and other related products intended to
support enteral feeding procedures. The Company’s business model includes the sale of the navigation system together with recurring
sales of disposable products used in connection with clinical procedures.
As
of the date of this Annual Report on Form 10-K, ENvue has established commercial engagements with multiple hospitals in the United States
for the evaluation, implementation, and clinical use of the ENvue System and related disposable enteral feeding tubes and accessories.
The Company’s commercialization strategy includes expanding its sales, marketing, and clinical education activities to support
increased adoption of the system and its associated disposable products across hospital intensive care units and other clinical settings
where enteral feeding procedures are performed.
Over
time, the Company intends to broaden the capabilities of its navigation platform and expand its product portfolio to address additional
clinical applications related to enteral access and other intrabody navigation procedures. In addition, ENvue may seek to expand its
commercialization activities into additional international markets, subject to obtaining applicable regulatory approvals and establishing
appropriate distribution and commercialization infrastructure.
ENvue
Products
| |
I. |
Enteral
Feeding - General Background |
Enteral
feeding is the most common method of providing liquid nutrition and certain types of medications to critically ill patients who are hospitalized
and require nutritional support, such as those on ventilators, post-surgery patients, patients with disabilities or conditions that prevent
them from eating fully or partially, and premature infants (Welch, Teresa D. “Nutrition options in critical care unit patients.”
Critical Care Nursing Clinics 30.1 (2018): 13-27 (“Welch 2018”); Milsom, S. A., et al. “Naso-enteric tube placement:
a review of methods to confirm tip location, global applicability and requirements.” World journal of surgery 39 (2015): 2243-2252
(“Milsom 2015”); de Aguilar-Nascimento, José Eduardo, and Kenneth A. Kudsk. “Use of small-bore feeding tubes:
successes and failures.” Current Opinion in Clinical Nutrition & Metabolic Care 10.3 (2007): 291-296 (“de Aguilar-Nascimento
and Kudsk 2007”); Koyfman, Leonid, et al. “The Placement of Post-pyloric Feeding Tubes Using DRX-Revolution Mobile X-Ray
System in an ICU. A Case Series.” The Journal of Critical Care Medicine 2.3 (2016): 131-134). The prevalence of malnutrition among
critically ill patients ranges from 30% to 50%, with some patients arriving at the hospital already malnourished and others potentially
developing malnutrition during hospitalization (Market & Markets; Barker, Lisa A., Belinda S. Gout, and Timothy C. Crowe. “Hospital
malnutrition: prevalence, identification and impact on patients and the healthcare system.” International journal of environmental
research and public health 8.2 (2011): 514-527; Wischmeyer, Paul E. “Malnutrition in the acutely ill patient: is it more than just
protein and energy?” South African Journal of Clinical Nutrition 24.3 (2011): S1-S7). It should be noted that there are critically
ill patients who, due to their medical condition, are unable to receive regular nutrition for many days.
Delays
in providing nutrition to patients can lead to a deterioration in their condition, as early insertion of the feeding tube and timely
provision of nutrition can, in most cases, reduce the severity of the illness, help preserve the integrity of the intestinal lining,
reduce infections and complications, improve gastrointestinal motility, and enhance immune response (Wang, Honggang, et al. “Early
enteral nutrition reduced postoperative ileus and improved the outcomes in patients with emergency intestinal surgery: results from a
propensity score analysis.” Int J Clin Exp Med 10.4 (2017): 7040-7048). Moreover, early provision of nutrition may improve the
patient’s condition and recovery rate, reduce possible complications, shorten the stay in intensive care, and even lower mortality
rates (Welch 2018). Consequently, early provision of nutrition may also result in significant cost savings for the hospital. Therefore,
in cases where regular nutrition cannot be provided to the patient and enteral feeding is required, it should be provided as soon as
possible (within 24-48 hours).
Enteral
nutrition is administered, among other methods, by inserting a feeding tube through the patient’s nose or mouth into the stomach
or small intestine (Tatsumi, Hiroomi. “Enteral tolerance in critically ill patients.” Journal of intensive care 7.1 (2019):
30 (“Tatsumi 2019”)). Each year, approximately 43 million nasogastric feeding tubes are inserted worldwide (about 14 million
of them in the United States) (Market & Markets). In general, according to FDA guidelines, a feeding tube inserted into a patient
should not remain in place for more than 30 days. However, hospitals tend to remove/replace the tube more frequently (for example, in
cases of tube blockage due to improperly dissolved medications or accidental disconnection of the tube by the patient).
Feeding
through a tube inserted through the patient’s nose or mouth directly into the patient’s small intestine (Post-Pyloric Feeding),
where nutrients are absorbed, requires more precise insertion of the tube, and offers several advantages over gastric feeding. Feeding
directly into the small intestine may reduce the risk of medical complications, involve a lower risk of gastric reflux and respiratory
infections and complications, provide higher caloric intake for the patient, and require a shorter stay in the intensive care unit (Welch
2018; Sajid, M. S., et al. “An integrated systematic review and meta-analysis of published randomized controlled trials evaluating
nasogastric against postpyloris (nasoduodenal and nasojejunal) feeding in critically ill patients admitted in intensive care unit.”
European journal of clinical nutrition 68.4 (2014): 424-432; Jiyong, Jing, et al. “Effect of gastric versus post-pyloric feeding
on the incidence of pneumonia in critically ill patients: observations from traditional and Bayesian random-effects meta-analysis.”
Clinical Nutrition 32.1 (2013): 8-15; Tatsumi 2019. As detailed below, the ENvue System is designed to facilitate the insertion of the
feeding tube through the patient’s nasal or oral route into the stomach or directly into the small intestine with accuracy and
efficiency.
Limitations
of Existing Alternative Enteral Feeding Methods
To
the best of ENvue’s knowledge, the most common method currently used for inserting feeding tubes through the nose or mouth is the
“blind” method, i.e., without visibility inside the patient’s body to facilitate accurate navigation and placement.
While for many years the “blind” insertion method was considered harmless, it has been found that this method can cause serious
and even fatal complications in patients (Market & Markets). Common complications among patients with feeding tubes inserted via
the “blind” method include incorrect insertion of the tube into the respiratory tract instead of the esophagus, aspiration
(entry of food, saliva, or stomach acids into the respiratory tract), lung collapse, sinus injuries, nosebleeds, and more (Rassias, Athos
J., Perry A. Ball, and Howard L. Corwin. “A prospective study of tracheopulmonary complications associated with the placement of
narrow-bore enteral feeding tubes.” Critical Care 2 (1998): 1-4; Prabhakaran, S., et al. “Nasoenteric tube complications.”
Scandinavian Journal of Surgery 101.3 (2012): 147-155 (“Prabhakaran 2012”)). According to studies, of the approximately 43
million feeding tubes inserted worldwide each year, about 1.72 million tubes are mistakenly inserted into patients’ lungs, and
of these, about 40% of patients suffered from pneumothorax (air accumulation in the chest cavity, impairing the breathing process) (de
Aguilar-Nascimento, Jose Eduardo, and Kenneth A. Kudsk. “Clinical costs of feeding tube placement.” Journal of Parenteral
and Enteral Nutrition 31.4 (2007): 269-273 (“de Aguilar-Nascimento 2007”); Burns, Suzanne M., et al. “Detection of
inadvertent airway intubation during gastric tube insertion: capnography versus a colorimetric carbon dioxide detector.” American
Journal of Critical Care 15.2 (2006): 188-195). Due to the high frequency of feeding tube use, experts believe that even a relatively
small percentage of cases where the feeding tube is incorrectly inserted could impact a very large number of people (Prabhakaran 2012).
Incorrect
insertion of the feeding tube into the lungs can have further serious consequences for the patient, including worsening of their medical
condition, which could lead to medical harm and even death, extended hospitalization, significant costs for the hospital, and legal claims
(de Aguilar-Nascimento 2007; Sparks, Dorothy A., et al. “Pulmonary complications of 9931 narrow-bore nasoenteric tubes during blind
placement: a critical review.” Journal of Parenteral and Enteral Nutrition 35.5 (2011): 625-629).
It
should be noted that due to technical failures and the prolonged time required for blind insertion of the feeding tube, attempts to insert
feeding tubes directly into the small intestine can result in delays in providing the necessary nutrition to the patient (as mentioned
above, providing early nutrition to the patient may improve their condition and prevent medical complications).
Given
the difficulties, risks, and possible complications associated with the blind insertion method of feeding tubes, as detailed above, the
duration of insertion, and the challenge of inserting it into the small intestine using this method, there is a noticeable trend towards
using alternative methods for inserting feeding tubes using technological or other means (Koopmann, Matthew C., et al. “A team-based
protocol and electromagnetic technology eliminate feeding tube placement complications.” Annals of surgery 253.2 (2011): 297-302
(“Koopman 2011”)), instead of relying on the blind insertion method, as detailed below.
There
are significant limitations in the existing alternative methods for inserting feeding tubes into patients and the various methods used
to verify that the blindly inserted feeding tube is located in the patient’s digestive system and not in the respiratory tract,
the main ones of which are detailed below.
Methods
for Verifying the Placement of a Blindly Inserted Feeding Tube
Among
the primary methods are measuring the distance of the tube from the insertion site, measuring the volume of aspirate, measuring the pH
level of the liquid aspirated from the tube (to check acidity levels to ensure it is gastric juices), checking the carbon dioxide level
of the air aspirated from the tube (to ensure the tube is not located in the lungs), and using an X-ray (fluoroscopy), with the latter
generally considered more accurate than the others (de Aguilar-Nascimento and Kudsk 2007; Milsom 2015). However, these methods may not
always identify errors accurately and in a timely manner, and they allow correction of incorrect insertion and placement of the tube
in the digestive system only after it has already been inserted into the lung, which may have caused pneumothorax or lung perforation
due to the insertion of the tube into the respiratory tract (Powers, Jan, et al. “Elimination of radiographic confirmation for
small-bowel feeding tubes in critical care.” American Journal of Critical Care 22.6 (2013): 521-527.). Furthermore, the X-ray method
has additional drawbacks, such as additional technical costs, prolonged time consumption, delayed patient nutrition, and radiation exposure.
Additionally, the pH measurement method has various limitations, such as respiratory burden and, primarily, inaccuracies due to medication
or other chemical treatment that affects the acidity level in the patient’s digestive system (Bourgault, Annette M., and Margo
A. Halm. “Feeding tube placement in adults: safe verification method for blindly inserted tubes.” American Journal of Critical
Care 18.1 (2009): 73-76).
The
core application of ENvue’s operations is the ENvue System, which is a system for monitoring and correctly positioning feeding
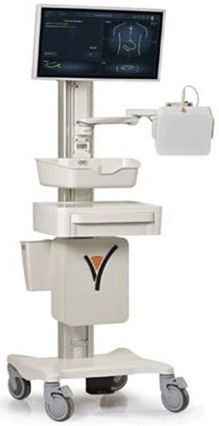
tubes in patients who require nutritional support during hospitalization. The system includes the main unit (the system body), disposable
(consumable) ENvue Feeding Tubes designed exclusively for use with the system, sensors, and additional components developed by ENvue.
These components, when used together, are intended to facilitate more efficient, faster, and safer insertion of the feeding tube into
the patient’s digestive system. The system uses electromagnetic waves transmitted to the upper torso of the patient, utilizing
sensors embedded in the dedicated feeding tube and sensors attached to the patient’s body during the procedure, enabling monitoring
and control of the feeding tube’s insertion path in an effort to facilitate proper placement into the GI tract bypassing the airways.
It should be noted that using the ENvue System will not guarantee the absence of medical errors or adverse events in connection with
a feeding tube placement. For example, five serious adverse events have been reported in connection with tubes inserted in patients’
lungs or pulmonary airway using the ENvue System. After investigation and review of placement files, ENvue believes the reported serious
adverse events were caused by user error.
Product
Components and Features
The
system components are described in greater detail below:
System
Body
The
system body includes several components, including a screen displaying the feeding tube’s position and an electromagnetic field
generator mounted on an adjustable arm. Before inserting the feeding tube and until the procedure is complete, the generator is positioned
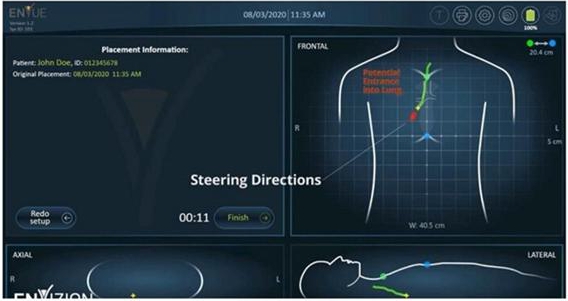
towards the patient’s chest and upper abdomen, emitting low-frequency electromagnetic waves throughout the procedure (see the
illustration below).
Once
the feeding tube is inserted, the passive electromagnetic sensor inside the tube enters the generator’s transmission area,
which detects the sensor’s movement and displays it graphically on the screen.
Reference
Sensor
An
external, reusable sensor connected to the system and attached to the patient’s body in the armpit area is used to reference
the feeding tube’s position within the patient’s body at any given moment. The reference sensor allows the system to
remain accurate even if the patient moves during the procedure (due to coughing, etc.).
Feeding
Tube (Disposable Component)
The
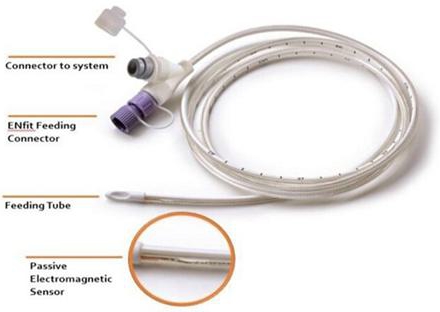
dedicated feeding tube developed by ENvue is designed specifically for use with the ENvue System and is intended for placement in
the stomach or small intestine. The feeding tube is single-use and features a dual connection: one for the nutrition source and another
for the ENvue System. |
|
During
the procedure, the other end of the tube is inserted through the nose into the patient’s body. A passive electromagnetic sensor
embedded in the tube allows the tube’s path within the patient’s body to be tracked on the system’s screen.
As
of now, ENvue’s feeding tube has been cleared in three different diameters: 8 Fr., 10 Fr., and 12 Fr.3
3
Fr. 1 = 0.3 mm.
ENvue
System Usage
The
ENvue System is used as follows: Throughout the feeding tube insertion procedure, the ENvue System emits electromagnetic waves toward
the patient’s upper body. Using the reference sensor connected to the system, the operator marks several anatomical points on the
patient’s upper body and attaches an additional location sensor to the side of the patient’s chest, allowing the system to
remain accurate even when the patient moves or coughs during the procedure. The operator then inserts the dedicated feeding tube for
the system through the patient’s nose or mouth into the esophagus and further into the digestive system (small intestine or stomach)
while viewing the tube’s path on the system’s screen from several angles and receiving real-time alerts if the system detects
the tube entering the patient’s airways, which is intended to enable the operator to immediately correct the tube’s insertion
path.
To
the best of ENvue’s knowledge, using the ENvue System allows the feeding tube insertion procedure to be completed within approximately
5-30 minutes on average, depending on the patient’s condition, the operator’s technical ability, and other factors. In comparison,
the time required for blind insertion of a feeding tube through the nose or mouth, based on ENvue’s estimate and medical research,
may take about 11-60 minutes (approximately 42 minutes on average) (Smithard, David, et al. “Electromagnetic sensor-guided enteral
access systems: a literature review.” Dysphagia 30 (2015): 275-285). Additionally, research shows that the time from blind insertion
of a feeding tube to the start of feeding the patient may take several hours, partly due to the need to verify the tube’s correct
placement in the digestive system using X-rays (Gray, Rebecca, et al. “Bedside electromagnetic-guided feeding tube placement: an
improvement over traditional placement technique?” Nutrition in Clinical Practice 22.4 (2007): 436-444).
Using
alternative methods as mentioned during the use of the ENvue System is not required by the FDA and is subject to the specific hospital’s
policy.
For
illustration purposes, below are diagrams demonstrating the use of the ENvue System:
Transmission
of electromagnetic waves by the system to the patient’s upper body throughout the procedure
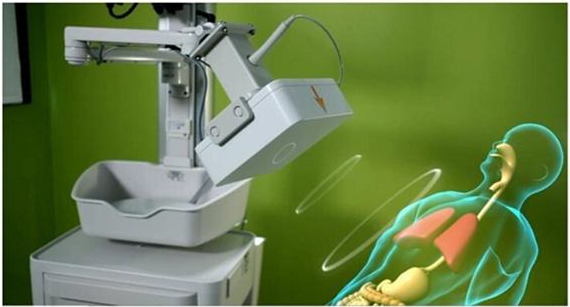
Real-time
visualization of the feeding tube’s insertion path within the patient’s body from multiple angles
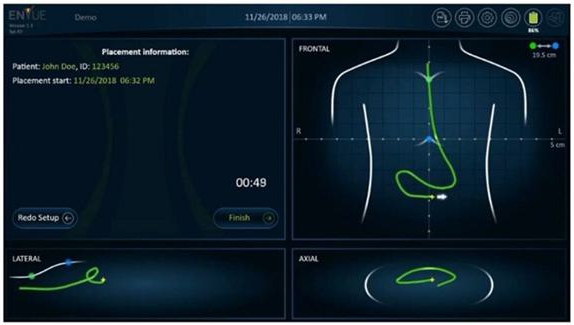
Receiving
an alert for the detection of improper insertion of the feeding tube
Marketing
Strategy
The
system, including the dedicated feeding tubes, is marketed to hospitals, and was designed and developed after ENvue received feedback
from healthcare professionals in the United States regarding their needs, which helped tailor the system to the market.
Using
the technology on which the system is based, it monitors the precise location of the feeding tube within the patient’s body, from
the moment it is inserted through the patient’s nose or mouth until it reaches the stomach or small intestine, and displays its
location in real-time through an imaging display of the patient’s body from several different angles on a screen. The display of
the patient’s body on the screen is made possible by using a reference sensor and marking anatomical landmarks on the patient’s
body at the beginning of the procedure.
In
cases where the system detects a deviation in the tube’s path towards the patient’s trachea, an immediate alert appears on
the screen. This allows the operator to correct the feeding tube’s insertion path immediately.
As
part of its operations ENvue has marketed the ENvue System to its customers and continuously supplied them with consumable feeding tubes,
which are designed for use exclusively with the system. At the beginning of 2020, ENvue began marketing the system and feeding tubes
to hospitals in the United States, following the FDA clearance received in February 2019 for marketing the product in this territory
for adults (aged 22 and older) only.
The
unique solution developed by ENvue as part of the ENvue System is intended to address, among other things, the risks and costs associated
with existing methods for inserting the feeding tube into the patient and the time required until the start of feeding due to delays
caused by the need to verify the tube’s placement in the patient, as described above. Using the ENvue System, including the dedicated
feeding tube developed by ENvue, it is possible to monitor the feeding tube insertion path into the patient and receive a real-time alert
if the tube is inserted into the patient’s respiratory tract. ENvue believes the system design may allow for accurate, reliable,
and efficient tube insertion for the patient and ease of use for the operator, which could potentially reduce the time required to insert
the feeding tube and thereby reducing the time until nutrition is provided to the patient.
4
The clinical trial lasted about a year, during which ENvue was required to obtain the consent of the patients or their family members
(depending on the patient’s medical condition) for participation in the trial.
Additionally,
ENvue believes using the system may minimize the risk of complications resulting from improper tube insertion and the associated costs
for the hospital, as well as shorten the patient’s hospitalization duration and prevent exposure to radiation from performing multiple
X-ray examinations to verify the tube’s placement. Furthermore, using the system screen that displays the patient’s body
dimensions, ENvue believes it is easier to properly insert the feeding tube into the patient’s small intestine on the first attempt,
which, as mentioned, is preferable to gastric tube insertion.
| |
III. |
Nutriseal
Nasogastric Aspiration Tube |
ENvue
also has another product in the field, a tube designed for enteral nutrition called the Nutriseal Nasogastric Aspiration Tube (NGAT).
This tube employs a sealing technique to prevent stomach acid from refluxing into the esophagus and to prevent aspiration of stomach
contents into the respiratory tract. NGAT has been cleared for marketing in the U.S. and was approved in the European Union; however,
as of now, ENvue is not manufacturing or marketing it, and the EU approval is currently not valid.
NGAT
is a feeding tube developed by Nutriseal Limited Partnership (the rights to which were transferred to ENvue shortly after its establishment
in 2017. NGAT is intended to serve as an enteral feeding tube for patients needing nutritional support and for other uses in hospitalized
patients. NGAT’s uniqueness compared to other feeding tubes lies in its sealing technique around the patient’s esophagus,
which can significantly reduce the risks associated with nasoenteral feeding (feeding through a tube inserted through the nose into the
stomach or small intestine), including esophageal reflux (the backflow of stomach acid up the esophagus) and aspiration (inhalation)
of stomach contents and food particles that may enter the lungs and cause severe health complications, including pneumonia.
NGAT
was developed and designed for use in various medical procedures, such as enteral feeding, gastric lavage, and gastric decompression,
while reducing the risk of esophageal reflux or aspiration. It should be noted that NGAT, in its current version, is intended for insertion
without a navigation system, but future iterations, if any, may be able to be used as a feeding tube connected to the ENvue System for
navigation during the tube insertion process, subject to applicable FDA clearance(s). Additionally, ENvue has developed other NGAT-b
components, for which, as of now, ENvue has not submitted applications for regulatory approval.
Product
Components and Features
NGAT
includes a feeding tube composed of a single central internal tube for delivering nutrients into the stomach and six internal suction
tubes surrounding the central internal tube (the “internal suction tubes”). NGAT contains small openings along its length,
which, after the tube is inserted into the patient’s body, are positioned along the esophagus and release low negative air pressure,
creating a suction action that causes the esophageal walls to contract inward, forming a seal around the tube that prevents stomach fluids
from refluxing into the esophagus and aspirating refluxed stomach fluids (the “aspiration mechanism”).
NGAT
is designed with two sealed suction areas located at the end of the tube inserted into the patient’s body, where stomach fluids
accumulate. The internal suction tubes are divided into two sets of three suction tubes each, with each set connected to a different
suction area. The operator can regulate the suction between the two sealed suction areas using a branched valve located at the end of
the tube that remains outside the patient’s body, allowing suction to be applied to one sealed suction area at a time.
NGAT
can be connected to standard hospital suction equipment, and its use does not require special equipment. This connection enables the
operation of the aspiration mechanism as well as performing gastric lavage procedures.
Marketing
Strategy
ENvue
may market NGAT in the U.S. for adult treatment only (aged 22 and above) (FDA clearance in the 510(k) pathway). NGAT also received the
European Union CE Mark for marketing in EU countries, which is not valid as of February 25, 2019, and several patents related to this
product have been registered.
ENvue
is not manufacturing or marketing NGAT as part of its business strategy to focus its operations and marketing efforts in the coming years
on the introduction and integration of the ENvue System into relevant markets. ENvue’s decision regarding the commercialization
of NGAT will be reviewed regularly by management and will be determined, among other factors, by the financial resources available to
ENvue, the pace of ENvue System adoption in the market, and the potential impact of various factors on ENvue’s operations (including
the risk factors to which ENvue is exposed). Therefore, there is currently no certainty regarding the timing of NGAT’s commercialization
by ENvue. It should be noted that NGAT can potentially be used with the ENvue System, subject to the necessary FDA clearance(s), and
ENvue may consider integrating NGAT within the ENvue System’s use and submitting an updated 510(k) notification to FDA if it decides
to commercially manufacture and market NGAT in the future.
ENvue
Feeding Tube and System for Use in Children and Preterm Infants
This
is a navigation system with dedicated feeding tubes of smaller diameters, designed for use in children and preterm infants. ENvue has
completed the initial product development process and will begin preparations for conducting a clinical trial as part of the FDA clearance
process.
Imaging
Navigation (ENvue Plus)
This
development allows the integration of medical imaging (fluoroscopy, MRI, CT) into the ENvue System, enabling real-time navigation of
the feeding tube based on the patient’s anatomical information. In January 2022, ENvue completed the product development process,
and is planning to initiate a clinical trial to assess the ENvue System’s capability to perform internal tube navigation based
on a chest X-ray image.
Peripherally
Inserted Central Catheter (PICC)
This
is a procedure for inserting a catheter into central blood vessels to ensure the catheter’s proper placement in the patient’s
blood vessels using the electromagnetic navigation technology of the ENvue System. This development is expected to allow ENvue System
users to perform PICC insertion with electromagnetic navigation on X-ray images, with real-time alerts from the ENvue System about incorrect
catheter placement in the patient’s body. Inserting a catheter into central blood vessels is essential for administering medications,
fluids, and nutrition and for taking continuous blood samples from hospitalized patients.
ENfit
Compatible Syringes
In
January 2026, ENvue announced the launch of ENfit compatible syringes designed for use with the ENvue System and enteral feeding tube
placements. ENfit is the internationally recognized connector standard (ISO 80369-3) developed to reduce the risk of misconnections between
enteral feeding devices and non-enteral lines. The ENfit compatible syringes are designed to work seamlessly with ENfit-compliant enteral
feeding tubes and connectors, supporting safe and accurate delivery of nutrition, hydration, and medications to patients requiring enteral
access. The Company believes the introduction of ENfit compatible syringes represents a meaningful expansion of its product portfolio
and supports its broader commercial strategy of providing a comprehensive enteral feeding solution to acute care hospitals in the United
States. In January 2026, ENvue signed a distribution agreement with U-Deliver, a U.S. based supplier of enteral feeding supplies, to
distribute ENvue’s ENFit compatible syringes through non-acute care channels nationwide. The syringes are available over the counter
in four sizes (2.5 mL, 5 mL, 10 mL, and 60 mL), meet ISO 80369-3 global standards, and are designed for reuse for up to seven days or
20 uses. Distribution is through U-Deliver’s website, Amazon storefront, and wholesale channels, targeting home care and long-term
care patients and caregivers. The agreement represents ENvue’s first commercial step into non-acute care channels, complementing
its existing acute care hospital business.
Robotic
Arm (ENvue Drive)
During
the year, the Company initiated development of a next-generation robotic platform within its ENvue Medical division, referred to as ENvue
Drive™, intended to automate aspects of electromagnetic navigation for enteral and vascular access procedures at the bedside.
The platform is being designed to integrate with the Company’s existing electromagnetic guidance technology used for enteral feeding
tube placement and is intended to support clinician alignment and positional stability during procedures. The system is currently in
the preclinical development phase and has not been submitted to the U.S. Food and Drug Administration (FDA) for regulatory clearance.
Development timelines and commercialization remain subject to engineering progress and applicable regulatory approvals.
Research
and Development
From
its founding, ENvue has engaged in the research and development of the ENvue System it developed—a system based on electromagnetic
navigation technology for inserting a feeding tube. In February 2019, ENvue received FDA clearance for the commercial marketing of the
product in the U.S. for adults (aged 22 and above) only.
ENvue’s
research and development activities have been focused mainly on product development and improvements and upgrades to various components
that make up the ENvue System to develop and improve performance.
As
part of the FDA clearance process for the system under the 510(k) pathway, ENvue was required to conduct a clinical trial following the
safety alert issued by the FDA regarding the use of the Cortrak*2 Enteral Access System, which is another device that, like the ENvue
System, is intended to facilitate enteral feeding tube placement, but was recalled due to serious adverse events resulting from misplaced
tubes in connection with the use of the system. During the clinical trial conducted by ENvue2, 58 feeding tube insertions
were performed on 57 subjects using the system, during which no feeding tubes were ultimately placed into the patient’s lung, and
no harm was caused to the patient’s lung during the procedure. In two cases, immediate correction of the tube insertion was performed
following the system’s alert of entry into the airways. Additionally, ENvue believes the trial results indicated ease of use of
the system and quick learning of how to operate it.
ENvue
Customers
ENvue’s
current and potential customers are hospitals in the U.S. As of 2023, there are approximately 5,222 hospitals in the U.S. that operate
intensive care units, which constitute the majority of ENvue’s potential customers (Fast Facts on U.S. Hospitals, 2024. American
Hospital Association. https://www.aha.org/statistics/fast-facts-us-hospitals). The primary users of ENvue’s products in these locations
are medical staff, usually nurses and clinical dietitians. As of the date hereof, ENvue’s customers include both hospitals in the
U.S. with which ENvue has signed direct sales agreements and hospitals within the hospital network with which ENvue has contracted under
the agreement detailed below.
ENvue
is in the stage of commercializing its products, and its sales activities in the U.S. were conducted directly with end customers. As
part of ENvue’s strategy to introduce the ENvue System into the U.S. market and expand its marketing efforts, ENvue may, in the
future, consider partnering with a distributor or strategic marketer, in addition to direct sales to customers in the U.S.
In
general, at large hospitals in the U.S., centralized procurement departments are responsible for the proposal submission process, contracting,
and negotiations for the purchase of all capital equipment. These departments emphasize economical and efficient operations. Typically,
a hospital procurement department consists of a procurement manager overseeing a team of senior and junior buyers. The process of purchasing
medical systems usually begins with the establishment of a Value Analysis Committee in the hospital, typically composed of physicians,
nurses, procurement agents, professional liability experts, supply chain management, and administrators. The committee coordinates discussions
with suppliers, visits sites where the systems are operated, and consults with colleagues from other hospitals. The main factors considered
by the procurement committee include (a) the hospital’s requirements based on a five-year forecast of patient needs (investment
horizon may vary between hospitals); (b) life cycle cost – total ownership cost; (c) economic considerations of cost recovery (cost
versus revenue); (d) performance, technical specifications, and physical data of the system; (e) workflow – capabilities, staff,
and output; (f) service, spare parts, and maintenance; (g) medical staff recommendations (quality of care).
ENvue
is not dependent on a single customer. The 4 customers comprise 18.6% of ENvue’s revenue.
2
The clinical trial lasted about a year, during which the Company was required to obtain the consent of the patients or their family
members (depending on the patient’s medical condition) for participation in the trial.
Description
of Key Terms of Engagement with End Customers
ENvue’s
agreements with its customers for the supply of the single-use-only ENvue Medical Enteral Feeding Tubes (EFTs) based on purchase orders
placed by the customer according to their needs, under terms outlined below. It should be noted that ENvue’s EFT is specifically
designed for use with the ENvue System. Per the FDA clearance for the ENvue System, which includes ENvue’s EFT, the system cannot
be used without this tube.
Negotiations
with ENvue’s customers are usually conducted by ENvue’s sales agents, following meetings with hospital procurement officials
and a demonstration (Demo) of the system, as well as evaluation by the medical staff through a few procedures of feeding tube insertion
in patients. Based on ENvue’s experience so far, the time from the demo to the receipt of a purchase order (PO) can take up to
6 months.
Under
ENvue’s agreements with customers for the purchase of the ENvue System, ENvue commits to manufacture, assemble, and supply the
system components to the customer according to schedules agreed upon by both parties for each engagement. Generally, the system is delivered
to the customer within a few weeks, and the feeding tubes are supplied within approximately one week from the customer’s purchase
order.
In
general, the consideration paid by the customer for the purchase of the system and feeding tubes is determined through negotiation between
the parties and according to ENvue’s discretion. Payment for the supply of the system and feeding tubes is typically made within
30 days of delivery, according to the specific engagement terms. It should be noted that in some cases, ENvue may provide the system
to the customer in exchange for replacing a competitor’s product owned by the customer, without any financial consideration (except
for the payment the customer will be required to make for the purchase of feeding tubes specifically for the system). The replaced competitor’s
product is used by ENvue, among other things, for training sessions it conducts for its customers on using the ENvue System instead of
the competitor’s product, as well as for ENvue’s research and development purposes.
The
agreement typically includes warranty periods for the system and the consumable feeding tubes, for periods defined in the agreement (generally
two years for the system and 30 days for the feeding tubes), during which ENvue commits to provide repair or replacement services for
defective components (defects) related to components manufactured by ENvue, within a timeframe agreed upon by the parties and according
to other terms set forth in the agreement. During the warranty period, ENvue may provide the customer with any necessary software and/or
hardware updates for the system, if any is needed for use of the system. In general, ENvue does not have a refund policy for its products
in most agreements.
Additionally,
the terms of engagement with the customer include standard cancellation clauses in appropriate circumstances (such as a material breach
of the agreement, company insolvency, etc.).
As
mentioned, customers who have purchased the ENvue System place ad-hoc orders with ENvue for the purchase of consumable feeding tubes,
according to the engagement terms outlined above. Customers issue a purchase order to ENvue, and in response, ENvue ships the products,
typically within a week. These orders are made according to the customer’s needs and generally on a monthly basis, with the consideration
determined by the price of the feeding tube agreed upon by the parties within the agreement.
EnVue
Sales and Marketing
ENvue
is continuously working to build awareness of its products among hospitals and the U.S. medical community in several ways, the main ones
being detailed below:
| |
● |
Conducting
Demonstrations for Potential Customers: As part of ENvue’s marketing activities, ENvue’s sales agents periodically
meet with hospital procurement officials, demonstrate (Demo) the system, and allow the medical staff to experience the system through
several procedures of feeding tube insertion in patients. |
| |
|
|
| |
● |
Engagements
with Group Purchasing Organizations (GPOs): A GPO is an entity that helps healthcare providers, such as hospitals, nursing
homes, and home health agencies, achieve savings and efficiency by aggregating purchasing volume and leveraging that to negotiate
discounts with manufacturers, distributors, and suppliers. ENvue believes that if it enters into agreements with such organizations,
it will allow for broader market penetration in relatively short timeframes. |
| |
|
|
| |
● |
Website
and Social Media: ENvue’s website provides information about ENvue and ways to contact it. Additionally, ENvue operates
several social media accounts, which include details about ENvue and its products, regular updates related to ENvue’s field
of activity, and the medical device market. |
| |
|
|
| |
● |
Press
Releases and Public Relations: ENvue publishes press releases related to agreements it has signed, new system deployments,
regulatory approvals received, and relevant milestones, such as significant capital and debt raisings. The announcements published
so far have generated media interest and have been covered in commercial media, national media, and technology publications. |
| |
|
|
| |
● |
Participation
in Events and Conferences: ENvue participates in selected events in the healthcare and technology industries to meet with
influencers and decision-makers in the field. |
ENvue
believes it is not dependent on any of its marketing channels.
ENvue
Market Opportunity and Trends
| |
I. |
Use
of Enteral Nutrition Means |
ENvue
operates in the market for enteral feeding devices for hospitalized patients needing nutritional support. The importance and use of enteral
nutrition means have been increasing over the last decade, partly due to their many advantages compared to traditional nutrition methods
in the market, such as parenteral nutrition (intravenous nutrition). Currently, enteral nutrition methods are widely used in many countries
worldwide. As of 2023, most enteral nutrition use was in North America (32.0%), Europe (29.0%), and Asia (24.0%) (“Enteral Feeding
Formulas Markt Size, Share, and Trends 2025 to 2034”; available at: https://www.precedenceresearch.com/enteral-feeding-formulas-market).
The
global enteral feeding devices market size was valued at $4.62 billion in 2025 (Enteral Feeding Devices Market Trends; Grand View
Research; available at: https://www.grandviewresearch.com/industry-analysis/enteral-feeding-devices-industry “Grand View Research”).
It is estimated that the market will grow at an annual rate (CAGR) (hereinafter: the “Growth Rate”) of approximately 5.86%
(about 4.4% in the USA) from 2024 to 2030, with the total market value expected to reach approximately $7.19 billion by 2030 (Grand View
Research).
Several
factors may drive the growth of the enteral nutrition market from 2024 to 2030, including rising healthcare costs, the increase in preterm
births, aging populations, the growing prevalence of chronic diseases such as diabetes, cancer, gastrointestinal diseases, and neurological
disorders, the increasing awareness of tube feeding, and improvements in healthcare systems in developing countries, among others.
However,
various factors, such as health risks, an increase in the number of malfunctions during patient feeding, and complications related to
tube feeding (such as faulty connections, tube disconnections, and infections), may limit the market’s growth. Additionally, incomplete
or no insurance coverage for using these means in countries where it is required (mainly developing countries), as well as a lack of
skilled medical personnel, are challenges to market growth.
Furthermore,
the rapid spread of the COVID-19 virus worldwide, especially the increase in morbidity in the USA, heightened the need to improve patient
nutrition, leading to increased demand for nasal enteral feeding means.
In
2023, hospitals were the primary users of enteral nutrition means (approximately 58.3% of global usage) (Grand View Research). The reasons
for this include the technological advancements of existing tube feeding methods, alongside the shift from intravenous nutrition to tube
feeding, which supports the growing use of these means in hospitals.
The
adult age group segment dominated the market with a revenue share of 91.3% in 2023 (Grand View Research). Projections indicate that in
the USA, this population is expected to remain the primary group using enteral nutrition means, with an expected growth rate of approximately
6.7% from 2020 to 2025 and a market value of approximately $3.9 billion by 2025.
| |
II. |
The
Use of Enteral Feeding Tubes |
Among
all enteral feeding methods, the market value of feeding tubes is the most dominant (approximately 45% in 2020). The use of enteral feeding
tubes includes, among other things, the insertion of an enteral feeding tube through the nose or mouth, as can be done using ENvue System.
It should be noted that most feeding tubes inserted through the mouth are intended for children and preterm infants. The ENvue System
is only cleared for use in adults, but ENvue believes that such clearance could potentially be expanded to include children and preterm
infants if ENvue is able to initiate and complete appropriately designed clinical studies that meet the endpoints necessary to demonstrate
that the system and its EFTs can be safely and effectively used in children and preterm infants and obtain the requisite FDA clearance
for such use.
According
to studies, approximately 43 million feeding tubes are inserted annually worldwide (Markets & Markets) through the nose, primarily
in North America (approximately 35%), Europe (approximately 28%), and Asia (approximately 24%), with an expected annual growth rate of
approximately 5.5%, 5.9%, and 9.1%, respectively, between 2020-2025.
Public
Awareness
In
recent years, there has been a growing trend in public awareness in Western countries, including the USA, regarding the importance of
using aids to ensure the proper insertion of feeding tubes into patients. This is mainly due to the increased awareness of the risks
associated with current insertion methods, such as patient lung injury, which can lead to lung collapse and even death. ENvue estimates
that this trend may increase the demand for its product in these countries due to its importance in minimizing the risks associated with
feeding tube insertion.
Awareness
in the Medical Community
The
medical community’s awareness of performing the feeding tube insertion procedure using ENvue’s product and the medical community’s
adoption of the solution offered by ENvue, instead of other methods and products in the market for performing the procedure, is significant
and crucial for ENvue’s success. Therefore, ENvue works with medical professionals in the USA to raise awareness among the medical
community. Additionally, ENvue works to raise awareness in this market, including through appearances at medical conferences, exhibitions,
participation, and conducting clinical studies for marketing purposes, as well as using various digital means.
Another
development in the general environment in which ENvue operates is the increasing use of the internet by medical professionals to obtain
information on new technologies and alternatives to existing methods for performing various medical procedures. Accordingly, ENvue works
to deepen public awareness and awareness among the medical community of the use of ENvue’s product as an alternative to existing
methods.
Medical
studies published in recent years regarding the risks associated with the use of existing methods for feeding tube insertion (mainly
the “blind” insertion method), as well as future studies on the subject, if published, may increase or decrease the demand
for ENvue’s product in the field in which it operates.
In
this context, it should be noted that the Patient Safety Movement organization6 published an article regarding the complications
and risks associated with the insertion of feeding tubes into patients, including ways to cope, guidelines, and recommendations for implementation
by hospital staff. The article emphasized the importance of proper feeding tube insertion in patients and identifying incorrect tube
insertion to ensure patient safety and the quality of medical care provided in the hospital, while reducing risks and preventing preventable
damage. The article outlines, among other things, guidelines, and actions to be taken by hospital medical staff to ensure proper placement
of feeding tubes in the patient’s body, including the limitations and risks associated with existing methods and technologies.
| 6 |
The Patient Safety Movement Organization is an American organization consisting of medical professionals from around the world, with the goal of preventing deaths caused by errors during hospital treatments.
See the link: https://patientsafetymovement.org/clinical/enteral-tube-safety/enteral-tube-safety-nasogastric-tube-ngt-placement-and-verification
And also: https://patientsafetyj.com/index.php/patientsaf/article/view/misplaced-nasogastric-tubes |
| |
III. |
Entry
Barries to the Target Market |
ENvue
estimates that there are significant entry barriers to the target market in which it operates. The main barriers to entry in ENvue’s
field of activity are as follows:
| |
● |
Scientifically
and Clinically Proven Technological Development - Pre-clinical and clinical work, which are usually essential conditions
for marketing a medical product, as well as the ability to ensure that a product that appears promising from a technological perspective
proves successful in the medical community, involve uncertainty, and create an entry barrier for competitors. |
| |
|
|
| |
● |
Regulatory
Constraints - The development, production, and sale of medical devices in the field typically require obtaining regulatory
approvals and meeting various standards depending on the country where the relevant activity is conducted, including approvals required
for conducting clinical trials in humans. A company that seeks to sell its products in a country where it does not have approval
for sale will often be required to invest significant resources, both time and money, to obtain the approval and the preliminary
processes. A company that is seeking to obtain similar regulatory approval or clearance to ENvue’s products or product candidates,
will need to meet the same or similar requirements and conditions that ENvue was required to meet, which may include conducting a
clinical trial, as ENvue was required to do to obtain FDA clearance under the 510(k) pathway for the commercial marketing of the
ENvue System in the U.S. |
| |
|
|
| |
● |
Intellectual
Property Protection - Products in the field are based on original technologies protected by patents or other intellectual
property rights in various countries. Intellectual property protections may prevent similar products from being marketed in relevant
countries for an extended period, potentially even decades. |
| |
|
|
|
|
● |
Initial
Capital and Knowledge - The development of products or processes in the field requires significant initial capital, appropriate
knowledge, and expertise. A product development project like ENvue’s products takes several years and requires extensive clinical,
biological, physiological, and chemical knowledge. A lack of funding or the required knowledge and expertise to conduct the research
and development could lead to the failure of the product’s development. |
| |
|
|
| |
● |
Skilled
Workforce - Developing, licensing, and producing products in the field requires professional and skilled personnel. A company
entering the field must recruit suitable personnel, and it may struggle to do so due to a lack of sufficient skilled and professional
workers. |
| |
|
|
| |
● |
Technological
Risk - Entering the field involves the risk that after significant investment of money and time, the developing company may
fail in developing the products, producing them, or obtaining the necessary approvals. Additionally, there is a risk that during
or after the completion of the development and licensing processes, it may become apparent that a competitor of the developing company
has developed a superior technology, giving them a competitive advantage. |
| |
|
|
| |
● |
Marketing,
Distribution, and Sales Capabilities - Companies operating in ENvue’s field of activity are required to establish,
finance, and maintain a sales and marketing infrastructure, whether through an internal team or by engaging with external distributors.
Each of the above options requires special and individual resources and connections in the field of activity, which constitutes a
barrier to entry for competitors. Additionally, there is a need for suitable marketing and distribution channels to handle institutional
bodies such as hospitals, which can compete against large companies operating in the field of ENvue’s products. |
| |
|
|
| |
● |
Rate
of Market Penetration - Penetrating the target market in the field of activity requires a long time, partly due to the entry
barriers described above. |
https://patientsafetymovement.org/clinical/enteral-tube-safety/enteral-tube-safety-nasogastric-tube-ngt-placement-and-verification
And also: https://patientsafetyj.com/index.php/patientsaf/article/view/misplaced-nasogastric-tubes
Competition
for ENvue System
ENvue
industry is competitive and has been evolving rapidly with the introduction of new products and technologies as well as the market activities
of industry participants. The ENvue System is indicated for use in adults 22 and over years of age to aid qualified operators in the
placement of the ENvue Medical Enteral Feeding Tube of 8 Fr, 10 Fr, and 12 Fr into the stomach or small intestine of adult patients requiring
enteral feeding. ENvue competes against other companies that have developed similar devices in the market for enteral feeding devices
for hospitalized patients needing nutritional support.
In
order to address the potential risks and complications associated with the “blind” insertion method of feeding tubes and
the drawbacks of standard methods for verifying the placement of the feeding tube, several products have been developed over the years
using technological tools to enable real-time monitoring of the feeding tube’s placement within the patient’s body. Based
on ENvue’s knowledge of the current landscape, there are two technological products on the market intended for use(s) similar to
that of the ENvue System: “IRIS Kangaroo Feeding Tube” (“IRIS Kangaroo”) and “Cortrak 2 Enteral Access
System” device (“Cortrak”). In addition, there are companies at various stages of developing feeding tubes with different
insertion methods that do not rely on intrabody navigation, and as of the date of this filing, ENvue does not consider them part
of its main competitors in the field of activity.
| |
|
ENvue
System |
|
IRIS
Kangaroo Feeding Tube |
|
Cortrak
2 Enteral Access System |
| |
|
ENvue |
|
Cardinal
Health |
|
Avanos
Medical3 |
| Product
Features and Usage |
|
A
navigation system based on electromagnetic technology used to assist in the efficient, safe, and quick insertion of a dedicated feeding
tube into patients, with real-time tracking of the tube’s insertion path and immediate alerts for incorrect insertion paths. |
|
A feeding tube platform incorporating camera-based visualization technology
intended to assist with tube advancement and placement The product has been in use since 2014. |
|
A
feeding tube with an electromagnetic component installed at the end, which emits electromagnetic
waves to a device placed on the patient, located in the Xiphoid Process area. This device
receives the waves and displays the tube’s position on a monitor. The product has been
in use for approximately 20 years.
|
3
To the best of ENvue’s knowledge, Avanos Medical acquired the product in 2016.
Market share data for products in this category is
not publicly available to our knowledge, and we therefore cannot reliably estimate comparative market share.
Competitive Considerations
IRIS Kangaroo utilizes camera-based visualization. As with any imaging-based
system, performance may depend on factors such as image quality, field of view, patient conditions, operator training, and workflow integration.
Certain customers may prefer alternative technologies depending on their staffing models, procedural preferences, and institutional requirements.
The
second product, Cortrak, is based on electromagnetic technology. This device displays the feeding tube insertion path in real-time
and allows the operator to navigate the tube into the digestive system. Preliminary studies have shown that inserting a feeding tube
in this way may reduce the need for X-rays (Hemington-Gorse, S. J., et al. “The use of the Cortrak Enteral Access
System™ for post-pyloric (PP) feeding tube placement in a Burns Intensive Care Unit.” Burns 37.2 (2011): 277-280;
Koopmann 2011). Customer preferences may vary based on installed base, familiarity, training pathways, clinical protocols,
purchasing agreements, and institutional experience. To the best of ENvue’s knowledge, the ENvue System is technologically
distinct from the Cortrak device in several respects, including: (1) performing a registration to the patient’s body that
allows for the display of the patient-specific chest contour according to individual dimensions on the system screen; (2) providing
a graphical and textual alert for feeding tube entry into the patient’s airway; (3) using the patient’s anatomical
landmarks for the navigation process; (4) additional sensors that enable accurate insertion of the feeding tube even if the patient
moves (without the need to place a device on the patient’s body); (5) a sensor embedded within the feeding tube; (6) three
simultaneous vies; and (7) responsive real time display of the tube tip pathway (40 image per second refresh rate) and more. The relevance of any particular feature may vary by customer, use environment, and clinical practice.
Regulatory and Market Events Involving Competing
Products
The competitive environment may be influenced by regulatory
actions, safety communications, recalls, published studies, professional guidance, and evolving standards of care affecting products in
our market.
In January 2018, the U.S. Food and Drug Administration
(“FDA”) issued a letter to healthcare providers regarding feeding tube placement systems following reports of cases in which
feeding tubes were placed into the lungs despite device indications of gastric placement. Among other things, the FDA stated that device
output should not be relied upon as the sole method of confirming feeding tube position. The communication did not prohibit the sale or
use of the affected product. Public safety communications of this nature may affect customer perceptions, purchasing criteria, training
requirements, and market adoption dynamics.4
In March 2022, Avanos Medical issued
field correction notices relating to the CORTRAK 2 Enteral Access System. On May 13, 2022, the FDA classified that action as a Class
I recall, the most serious type of recall classification, noting that incorrect placement of nasogastric or nasoenteric tubes could lead
to serious injury or death. We cannot predict the extent to which any such event involving a competing product may benefit us, harm us,
or have no material effect on our business. Competing products may remain available and continue to be used notwithstanding such events.5
4 U.S.
Food and Drugs Administration: Feeding Tube Placement Systems: Letter to Health Care Providers (January 2018), available at: https://www.fda.gov/medical-devices/letters-health-care-providers/feeding-tube-placement-systems-letter-health-care-providers
5 U.S.
Food and Drug Administration, Medical Device Recall Database, CORTRAK 2 Enteral Access System, Class I Recall Classification posted May
13, 2022, available at:https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfres/res.cfm?id=193098
Additionally,
on March 21, 2022, Avanos Medical initiated a voluntary recall of the Cortrak device, issuing a field correction notice to customers.
The FDA subsequently classified the recall as Class I — the most serious type of recall — on May 13, 2022, noting that incorrect
placement of nasogastric or nasoenteric tubes using the device could lead to serious injury or death.
Pricing and Economic Competition
Traditional blind-placement methods may involve lower
direct product costs than technology-assisted placement systems. However, customers evaluate products using a range of factors beyond
unit price, including workflow efficiency, staffing requirements, training burden, confirmation procedures, clinical confidence, patient
safety considerations, total cost of care, and institutional protocols.
Competing technology-assisted systems may be priced
similarly to, above, or below our offerings depending on configuration, purchasing volume, contracting arrangements, and geography. Pricing
pressure, bundled purchasing strategies, group purchasing contracts, distributor leverage, and competitor discounting could adversely
affect our ability to grow revenue or maintain margins.
ENvue’s
Main Strategies for Dealing with Competition
EnVue
has historically dealt with competition in its market by differentiating and developing the technology of its products, developing a
training model for customers, investing in the deployment of a service sales network, and an effective marketing strategy. Additionally,
EnVue worked to protect its intellectual property by registering its intellectual property rights in countries where it identifies potential
activities, in order to maintain the competitive advantage of its products in the field of activity.
EnVue
estimates that the ENvue System it developed provides it with a competitive advantage over other products available in the market, as
well as the quality of the products and services it provides, and its intellectual property protected by patents. The factors strengthening
ENvue’s competitive position are described in further detail below:
| (1) |
Technological
Capability and Unique Operating Method of ENvue’s Product: The ENvue System has an advantage over competing products by
providing a stable real-time image of the patient’s body, regardless of patient and/or device movements. It also does not require
special expertise and can be operated by a trained care provider. These advantages pose a challenge for competitors who struggle
to achieve the same level of reliability and stability in products and services in the field of activity, including the unique and
effective electromagnetic navigation method, which is partly patented. |
| (2) |
Significant
Technological Improvement Compared to Existing Methods: We believe our platform addresses important customer needs relating to
bedside workflow, procedural efficiency, and confidence during placement procedures. Actual outcomes may vary by institution, operator
training, patient population, and clinical practice. |
| (3) |
Skilled
and Experienced Workforce: Our personnel bring experience across engineering, operations, commercialization,
regulatory affairs, and customer support. We believe this cross-functional capability supports continued growth and execution. |
| (4) |
Intellectual
Property: We maintain patents, trademarks, know-how, and other proprietary rights
in multiple jurisdictions. We believe our intellectual property portfolio supports our competitive positioning, although there can be
no assurance that such protections will prevent competition or provide a material commercial advantage. |
Nano
OpCo’s Business
Nano
OpCo’s primary products, which are in various stages of clinical and market development, currently consist of:
| |
● |
PainShield,
a patch based therapeutic ultrasound technology designed to treat pain, muscle spasms and
joint contractures by delivering localized ultrasound energy that promotes pain relief and
supports soft tissue healing in targeted areas, including PainShield MD, a single use patch-based
device utilizing this technology.
|
| |
● |
UroShield,
an ultrasound-based product that is designed to prevent bacterial colonization and biofilm in urinary catheters, increase antibiotic
efficacy and decrease pain and discomfort associated with urinary catheter use. It is currently being marketed in Europe under a
CE mark. |
Each
of PainShield and UroShield employs a small disposable transducer that transmits low frequency, low intensity ultrasound acoustic waves
designed to support tissue repair and regeneration of musculoskeletal and vascular structures. The technology is also intended to decrease
biofilm formation, reduce catheter blockage, and alleviate pain associated with urinary catheters, while potentially reducing the incidence
of catheter associated urinary tract infections.
Through
their size, effectiveness and ease of use, these products are intended to eliminate the need for technicians and medical personnel to
manually administer ultrasound treatment using large transducers, thereby promoting patient independence and enabling more cost effective
home based care.
PainShield
MD is currently cleared for marketing in the United States by the FDA. Both PainShield and UroShield have CE Mark approval in the European
Union and hold regulatory certification permitting commercial sale in Israel.
In
the United States, PainShield and UroShield require a prescription from a licensed healthcare practitioner.
Insurance
Coverage and Reimbursement
In
addition to the need to obtain regulatory approvals, we anticipate that sales volumes and prices of NanoVibronix’s UroShield and
PainShield, products will depend in large part on the availability of insurance coverage and reimbursement from third party payers. Third
party payers include governmental programs such as Medicare and Medicaid and the Veterans Health Care network of facilities in the United
States, private insurance plans and workers’ compensation plans. Effective as of January 2020, the U.S. Centers for Medicare and
Medicaid Services (“CMS”) approved our PainShield for reimbursement for Medicare beneficiaries on a national basis. However,
PainShield was not assigned a reimbursement value from CMS. The Company was denied reimbursement in September 2022 due to a lack of “life-cycle”
testing. We are currently evaluating whether to resubmit another application to CMS.
With
respect to UroShield, which may be used in a clinical and home setting, we currently have reimbursement in the United Kingdom - The NHS
Drug Tariff Part IX establishes a direct, prescription-based reimbursement pathway, for the UroShield® Kit (November 2025), which
is used in conjunction with the UroShield® Actuator that was previously included in the NHS Drug Tariff Part IX in the UK in 2023, and throughout the VA system. We are seeking reimbursement codes for use of our products in the markets in which we have regulatory
authority, including the United States, to sell such products. Our current ongoing research and planned research may facilitate our ability
to obtain reimbursement codes, but there is no guarantee that we will be successful in obtaining such codes quickly, or at all.
Nano
OpCo’s Business Model
All
of Nano OpCo’s products consist of a reusable controller device and a disposable component, which includes a transducer, and in
the case of PainShield, a 30-day supply of adhering patches. The components are purchased by either the distributor or end user for
use in any of the intended applications. Once the controller is purchased by the end user, recurring revenue will be realized by purchases
of replacement disposables to the extent that the end user continues treatment with our product.
Nano
OpCo’s products are intended to be distributed directly by either the dedicated sales force, independent distributors, and potential
licensees. Distributor cost is discounted to account for their intended margins, based upon purchase volumes and/or periodic purchase
commitments, with the disposable transducer sold and distributed in the same fashion. We currently have an established distributor network
and are implementing certain criteria within such network to ensure the appropriate assignment of a distributor or licensee.
Nano
OpCo’s business plan continues to focus on these types of transactions/agreements. We continue to focus on the foundational aspects
of each respective product, including the design and performance of each, the reimbursement, regulatory status, and quality control,
in order to strengthen our position with prospective partners.
Ultrasound
Technology and Nano OpCo’s Products
As
noted above, Nano OpCo’s primary products are based on the use of low frequency ultrasound, which delivers energy through mechanical
vibrations in the form of sound waves. Ultrasound has long been used in physical therapy, physical medicine, rehabilitation and sports
medicine.
Our
proprietary PainShield technology consists of a small, thin (1 millimeter) transducer that is capable of transmitting ultrasonic acoustic
waves onto treatment surfaces with a radius of up to 10 centimeters beyond the transducer. This technology allows us to treat pain by
securing our transducers to the skin with a separate adhesive patch, thereby eliminating the need for technicians and medical personnel
to manually administer ultrasound therapy, which should reduce the cost of therapy. Moreover, we believe that, based upon the body of
evidence, the delivery of ultrasound through our portable devices may provide a competitive advantage over other existing therapies marketed
for similar intended use(s) (e.g., to treat pain associated with muscle, tendon, and contractures), as our technology is positioned to
directly target the affected areas of the body within the scope of the applicable FDA clearance.
While
there are currently a number of products on the market that treat pain through ultrasound therapy, we believe that our products may be
preferable in certain instances because they are portable, without the requirement to be plugged into an outlet and they have a frequency
of 100kHz (in contrast to other devices, which have a frequency of closer to 1MHz and above), which means our products, when functioning
as intended and in accordance with applicable design specifications, should not produce excessive heat that can damage tissue. Therefore,
our products (i) can be self-administered by the patient without the need to be moved about the treated area by the patient or a clinician,
(ii) can be applied for a significantly longer period without the risk of tissue damage and (iii) do not require the use of gel. We are
also aware of one product, the SAM® Sport family of products, which received FDA approval after PainShield and has CE Mark approval,
marketed by ZetrOZ, Inc., that we understand may eliminate certain of these requirements and limitations, namely the requirement to be
plugged in, the need for movement around the treated area and the relatively short safe treatment period. However, we understand that
this product does not generate surface acoustic waves as our products do, which means that the treatment area is generally limited to
that under the transducer, that the use of transmission gel is still required, and that the transducer thickness is significantly greater
than ours (approximately 1.5 cm). It is also our understanding that the FDA has issued multiple contraindications for SAM® Sport,
which do not apply to the PainShield product.
There
has been an article published in 2019 on SAM® Sport4 regarding clinical evidence demonstrating that ultrasound dose timing (i.e.
daily treatment) and duration significantly impact benefits and treatment results. We are aware of a prospective randomized, double-blinded,
placebo-controlled study on the effects of the long-duration low-intensity ultrasound treatment using SAM® Sport4 suggesting that
ultrasound may be used as a conservative non-pharmaceutical and non-invasive treatment option for patients with knee osteoarthritis.
In
general, ultrasound offers the benefits by increasing local blood circulation, increasing vascular wall permeability, promoting protein
secretion, promoting enzymatic reactions, accelerating nitric oxide production, promoting angiogenesis (the formation of new blood vessels
from pre-existing vessels) and promoting fibroblast proliferation (fibroblasts are a type of cell that play a critical role in soft tissue
healing). We believe that the body of evidence, and the positive therapeutic effect that ultrasound has for various indications, potentially
provides for future product development opportunities for us.
| Conventional
Ultrasound |
PainShield
Ultrasound |
 |
 |
Traditional
ultrasound device and our portable ultrasound patch-based device and a comparison of their energy distribution, where the X-axis represents
treatment surface, and the Y-axis represents ultrasound energy penetration depth within tissue.
In
a comparison of a traditional ultrasound device and our portable ultrasound patch-based device, the bulk wave conventional ultrasound
machines with handheld transducers distribute the energy deeply into the body, as shown above in diagram (A) on the left. In comparison,
our device distributes the energy on the surface, as shown in diagram (B), thereby meaningfully increasing the treatment area. Our transducers
may also be incorporated into treatment patches, including patches that are designed to deliver medicine and other compounds through
the skin. The generation and delivery of low frequency ultrasound over a period of time to a specific area has been termed “targeted
slow-release ultrasound”. We believe that this delivery method of ultrasound may be comparable to that of slow-release medication
in the pharmaceutical industry. This “targeted slow-release” capability is intended to allow for more frequent targeting
of the intended treatment area and thus may result in a more effective therapeutic response.
Micro
Vibrations Technology and Nano OpCo’s Products
In
a 2007 study, increase in mean blood flow to the calf was higher in the vibration group than the placebo group. Improvements in local
blood flow may be beneficial in the therapeutic alleviation of pain or other symptoms resulting from acute or chronic injuries (C. Button
et al., “The effect of multidirectional mechanical vibration on peripheral circulation of humans”, University of Otago New
Zealand, Clinical Physiology and functional Imaging, 2007 27, p211-216). A study on the effect of whole body vibration on lower extremity
skin blood flow suggests, that short duration vibration alone significantly increases lower extremity skin blood flow, doubling skin
blood flow for a minimum of 10 minutes following treatment (Lohman et al., “The effect of whole body vibration on lower extremity
skin blood flow in normal subjects”, Department of Physical Therapy, Loma Linda university, USA, Med Sci Monit, 2007; 13(2) 71-76).
Vibration has also been shown to stimulate angiogenesis and growth factors such as vascular endothelial growth factor (Suhr F et al.,
“Effects of short-term vibration and hypoxia during high intensity cycling exercise on circulating level of angiogenic regulators
in humans”, J Appl Physiol, 2007, 103:474-483, Yue Z. et al., “On the cardiovascular effects of whole-body vibration I. Longitudinal
effects: hydrodynamic analysis”, Studies Appl Math, 2007, 119:95-109).
Relative
to soft tissue repair, it is well established that increasing blood flow to the wound and peri-wound area helps accelerate the healing
of ischemic wounds. Micro-vibrations applied on the skin tissue increase local blood flow and oxygen delivery to the wound area and stimulate
angiogenesis and growth factors that are helpful for the wound healing process. Vibration therapy has been found to stimulate blood flow
due to mechanical stresses of endothelial cells resulting in increased production of nitric oxide and vasodilation, as well as increase
soft tissue and skin circulation. (Maloney-Hinds et al., “The Role of Nitric Oxide in Skin Blood Flow Increases due to vibration
in healthy adults and adults with type 2 diabetes,” School of Medicine, Loma Linda University. Ca. Diabetes Technology & Therapeutics,
2009 p. 39-43). In addition, micro vibrations induce skin surface nerve axon reflex and type IIa muscle fibers contraction rates, resulting
in vasodilation (Nakagami et al., “Effect of vibration on skin blood flow in an in vivo microcirculatory model”, The University
of Tokyo, Bio-Science Trends 2007; 1 (3): 161-166). Ten minutes of vibration therapy with laser doppler revealed a consistent increase
in water content of the upper dermis (TJ Ryan et al., “The effect of mechanical forces (vibration or external compression) on the
dermal water content of the upper dermis and epidermis, assessed by high frequency ultrasound”, Oxford Wound Healing Institute,
Journal of Tissue Viability, 2001. Of import with respect to diabetic wounds, in which a prolonged inflammatory phase occurs, vibration
vasodilation has generated an indirect anti-inflammatory action, mainly by suppression of nuclear factor-kβ, the key gene for inflammatory
mediators (Sackner, M.A., “Nitric Oxide is released into circulation with whole-body, periodic acceleration”, Chest 2005;127;30-39).
Urinary
catheter usage is associated with pain and discomfort caused by the friction between the catheter surface and the urethral tissue. Generally,
this friction is treated by applying lubricating gels and low friction catheter coatings. These methods are effective for a short term
during the catheter insertion as the lubricating gel is quickly absorbed into the surrounding tissue and loses its effect and the catheter
coatings lose their lubricity within a few days, as the coating is covered by a thin film of mucous.
Our
UroShield product provides vibrations along the surface of the urinary catheter that is in contact with urethral tissue. We believe that
these vibrations create a continuous acoustic lubrication effect along the surface of the indwelling catheter that is in contact with
the surrounding tissue, thus reducing catheter-tissue contact time, which may lessen trauma from urethra abrasion and adhesion. We have
also shown in animals and in humans that the micro-vibration technology can reduce the level of biofilm formation on urinary catheters.
Nano
OpCo’s Products
Product
Design, Packaging, Identity
We
currently complete assembly in our facilities in Israel and planning to move manufacturing to a Contract Manufacturer located in the
US. Even though our ability to assemble our products has not been affected by the current political environment, we cannot predict if
future events may cause delays. Our 2025 production run established an ample supply of devices and monthly disposable kits. The completed
products can be used as a platform for either PainShield or UroShield.
UroShield
UroShield
is intended to prevent bacterial colonization and biofilm formation, increase antibiotic efficacy in the catheter lumen and decrease
pain and discomfort associated with urinary catheter use. It is designed to be used with any type of indwelling urinary catheter regardless
of the material or coating. Use of the device is contraindicated for use while there is an active UTI. We believe that UroShield may
be the first medical device on the market that attempts to simultaneously address all of the aforementioned catheter-related issues.
UroShield is similar in design to PainShield, in that it uses a driver unit that produces low frequency, low intensity ultrasound. The
driver unit connects to a disposable transducer that is clipped onto the external portion of the catheter to deliver ultrasound therapy
to all catheter surfaces as well as the tissue surrounding the catheter.
Picture
of UroShield with actuator
Clinical
studies of the UroShield system have supported the following advantageous effects:
| |
● |
Prevention
or Reduction of Biofilm. The low frequency ultrasound generated by UroShield has been shown to decrease adherence of bacteria
to catheter surfaces, thereby reducing biofilm. Biofilm is the complex matrix required for bacteria to grow and cause infection.
See the discussion of our Heidelberg 1 trial below. |
| |
|
|
| |
● |
Decreased
Catheter Associated Pain and Discomfort. We believe that UroShield creates an acoustic envelope on the surfaces of the catheter,
which decreases friction and tissue trauma, pain and discomfort caused by the catheter. In addition, in vivo (rabbit) studies have
shown the tissue in contact with the catheter remains healthier and less traumatized as a result of the application of low frequency
and low intensity ultrasound (Applebaum I, et.al., “The Effect of Acoustic Energy Induced By UroShield on Foley Catheter Related
Trauma and Inflammation in a Rabbit Model” Department of Urology, Shaarey Zedek Medical Center and the Hadassah Hebrew University
Medical School). |
| |
|
|
| |
● |
Acoustically
Augmented Antibiotic Therapy. Antibiotic resistance in biofilm bacteria is a well-known phenomenon. Although it has been known
that ultrasound can increase antibiotic efficacy in in-vitro models, we do not believe that there has been a practical ultrasound-based
medical device that was able to augment antibiotic efficacy in the clinical setting. In a clinical study, UroShield technology has
been shown to eradicate biofilm-residing bacteria by greater than 85% when applied simultaneously with an antibiotic in three clinically
relevant species, escherichia coli, staphylococcus epidermidis and pseudomonas aeruginosa (Banin E, et al., “Surface acoustic
waves increase the susceptibility of Pseudomonas aeruginosa biofilms to antibiotic treatment,” Biofouling, August 2011; we
supplied devices for this study, but had no further involvement with it). |
| |
|
|
| |
● |
Preservation
of the Patency of Catheters. We believe that low frequency ultrasound applied to catheters will add an anti-clogging effect and
will preserve patency of catheters. This effect is achieved by ultrasound waves creating an acoustic layer on the inner lumen of
the urinary catheter, thereby preventing adherence of biological material and biofilm formation. We believe that this anti-clogging
benefit will help prevent local infection and sepsis secondary to catheter obstruction. |
In
November 2025 the UroShield® Kit has been added to the UK National Health Service (NHS) Drug Tariff Part IX, enabling nationwide
prescription reimbursement across the UK.
The
NHS Drug Tariff Part IX establishes a direct, prescription-based reimbursement pathway, significantly expanding access for the UroShield®
Kit, which is used in conjunction with the UroShield® Actuator that was previously included in the NHS Drug Tariff Part IX in the
UK in 2023, across community and hospital care settings.
UroShield
will be available to all patients who need the device with full clinical support, through the NHS supply chain. It represents a significant
opportunity for us to expand distribution of UroShield as it will now be made available to all clinicians and their patients through
the NHS organization’s own supply channel. NHS Supply Chain manages the sourcing, delivery and supply of healthcare products and
services for NHS trusts and healthcare organizations across England and Wales. The organization processes more than eight million orders
per year across 94,000 order points and 17,465 locations serving as an integral part of the national healthcare system in the U.K.
A
urinary tract infection (UTI) is defined by the Centers for Disease Control and Prevention (CDC) as an infection involving any part of
the urinary system, including the urethra, bladder, ureters, and kidney (CDC, 2024). UTIs remain the most common type of healthcare-associated
infection (HAI) reported to the National Healthcare Safety Network (NHSN), and catheter-associated UTI (CAUTI) continues to account for
the largest share of HAIs in both hospital and long-term care settings.
Among
UTIs acquired in hospital settings, approximately 75% are linked to an indwelling urinary catheter. An estimated 15–25% of hospitalized
patients will have an indwelling catheter at some point during their stay, and approximately 7% of nursing home residents are managed
via long-term catheterization (CDC, 2024).
| |
2. |
Current
Epidemiology (2023–2025) |
| Metric | |
Current
Data (2023–2025) |
| CAUTI
share of all HAIs | |
~26–30% of all
reported HAIs (NHSN 2023) |
| ICU
CAUTI rate | |
~0.9–1.4 per
1,000 catheter-days (NHSN 2023) |
| Non-ICU
CAUTI rate | |
~0.5–0.9 per
1,000 catheter-days (NHSN 2023) |
| Patients
with catheter during stay | |
15–25% of all
hospital admissions |
| Nursing
home catheter prevalence | |
~7% of residents (long-term
catheterization) |
| Daily
bacteriuria risk (catheterized) | |
3–10% per day;
~25% at 7 days |
| Bacteriuria
at >30 days catheterization | |
Virtually 100% |
| CAUTI-related
bacteremia (acute care) | |
~17–20% of healthcare-acquired
bacteremia |
| CAUTI-related
bacteremia (long-term care) | |
~50% of healthcare-acquired
bacteremia |
CAUTI
occurs because urethral catheters inoculate organisms into the bladder and promote colonization by providing a surface for biofilm formation
and causing mucosal irritation (Maki & Tambyah, 2001). The extraluminal route (migration of organisms along the catheter-meatus interface)
is responsible for the majority of early infections, while the intraluminal route becomes more significant with longer catheter dwell
time.
The
most common causative organisms identified in recent NHSN surveillance (2015–2022) include (Weiner-Lastinger et al., 2021):
| |
● |
Escherichia
coli (~21%) |
| |
● |
Candida
spp. (~18–21%) |
| |
● |
Klebsiella
pneumoniae (~11%) |
| |
● |
Enterococcus
spp. (~10%) |
| |
● |
Pseudomonas
aeruginosa (~8%) |
Of
note, the proportion of antimicrobial-resistant pathogens causing CAUTI has risen, with ESBL-producing organisms and fluconazole-resistant
Candida species now representing a growing clinical challenge (Weiner-Lastinger et al., 2021).
4.
Economic and Healthcare Burden (Updated)
4.1
Cost per CAUTI Episode
The
economic burden of CAUTI has been re-evaluated in multiple recent analyses. The cost per CAUTI episode attributable to the catheter ranges
broadly depending on complexity, but current estimates are (AHRQ, 2023; Mitchell et al., 2023):
| |
● |
Simple
CAUTI (uncomplicated): $896–$2,836 per episode in attributable costs |
| |
● |
CAUTI
with secondary bacteremia: $13,793–$32,000+ per episode, reflecting increased LOS and treatment costs |
| |
● |
Total
annual US burden: estimated at $340 million to $450 million annually |
4.2
Global Catheter Market (2025 Data)
The
global urinary catheter market was valued at approximately USD 4.5–5.2 billion in 2023 and is projected to reach USD 7.0–8.5
billion by 2030, growing at a CAGR of approximately 6.5–8.2% (Grand View Research, 2023-2024). This revision reflects updated segmentation
methodology separating urinary catheters from the broader catheter market reported in earlier analyses.
In
the United States, approximately 30 million urinary catheterization procedures occur annually (including both intermittent and indwelling),
with Foley catheter usage estimated at 20–25 million units per year. Global Foley catheter sales are estimated at 80–100
million units annually (Grand View Research, 2023-2024).
Key
growth drivers include the aging global population, rising prevalence of urological disorders and diabetes, expanded ambulatory surgical
procedures, and growing adoption of antimicrobial/anti-biofilm catheters.
5.
Current Prevention Guidelines (2023–2025)
The
most current CAUTI prevention guidance comes from a 2023 joint update by the Society for Healthcare Epidemiology of America (SHEA), the
Infectious Diseases Society of America (IDSA), and the Association for Professionals in Infection Control and Epidemiology (APIC) (Lo
et al., 2023). Key recommendations include:
5.1
Appropriate Catheter Use (Indication-Based)
| |
● |
Insert
catheters only for appropriate indications; avoid use for incontinence management alone. |
| |
● |
Implement
real-time nursing-driven catheter removal protocols (“CAUTI bundles”). |
| |
● |
Consider
alternatives: condom/external catheters for males, intermittent catheterization, and absorbent products. |
5.2
Insertion and Maintenance
| |
● |
Maintain
a sterile, closed drainage system at all times. |
| |
● |
Ensure
unobstructed urine flow; keep bag below bladder level. |
| |
● |
Perform
hand hygiene before and after any catheter manipulation. |
| |
● |
Routine
catheter replacement is NOT recommended on a fixed schedule; replace only if obstruction or contamination occurs. |
5.3
Technology Interventions
| |
● |
Antimicrobial
catheters (silver-alloy or nitrofurazone-coated): recommended for short-term use in high-risk settings, though evidence for long-term
benefit is mixed (Lo et al., 2023). |
| |
● |
Electronic
reminder and stop-order systems are strongly recommended as system-level interventions to reduce unnecessary catheter days (Lo et
al., 2023). |
| |
● |
Novel
anti-biofilm coatings (e.g., hydrophilic polymer coatings, bacteriophage-based coatings) are in clinical trial phases as of 2024–2025. |
6.
CMS / Reimbursement Policy (Current Status)
Since
October 1, 2008, the Centers for Medicare & Medicaid Services (CMS) has not reimbursed hospitals for the additional costs associated
with CAUTI when it was not present on admission (POA). CAUTI is classified as a Hospital-Acquired Condition (HAC) (CMS, 2025).
As
of 2025, additional enforcement mechanisms are active:
| |
● |
HAC
Reduction Program (HACRP): Hospitals in the worst-performing quartile for HAC rates (including CAUTI) receive a 1% reduction in Medicare
payments. This program has been in place since FY2015 and continues to be enforced annually (CMS, 2025). |
| |
● |
Hospital
Value-Based Purchasing (HVBP): CAUTI rates are a scored domain, directly affecting hospital reimbursement rates (CMS, 2025). |
| |
● |
CMS
Star Ratings: CAUTI rates factor into CMS Hospital Compare star ratings, creating reputational and referral-based financial incentives
beyond direct reimbursement. |
Competition
for UroShield
Several
types of products have been introduced to address the growing problem of catheter-acquired infection and biofilm formation on catheter
surfaces. Manufacturers offer antibiotic-coated and antiseptic-impregnated catheters. In addition, manufacturers have produced silver-coated
catheters, which have been shown in small studies to delay bacteriuria for about two to four days. However, larger studies did not corroborate
this result; on the contrary, silver hydrogel was associated with overgrowth of gram positive bacteria in the urine (Riley DK, Classen
DC, “A large randomized clinical trial of a silver-impregnated urinary catheter: lack of efficacy and staphylococcal superinfection,”
Am. J. Med. 1995 April; 98(4):349-56).
UroShield
has been designed to be added to any type of catheter, including Foley catheters and silver-coated catheters, to improve a catheter’s
infection prevention performance. However, in the United States, we do not have the requisite regulatory authorization to market UroShield
for such use, as we have not yet obtained FDA clearance or approval for UroShield. UroShield is not intended to replace any existing
products or technologies, but instead is intended to assist these existing products or technologies in preventing catheter-acquired urinary
injury and catheter associated complications.
Regulatory
Strategy
UroShield
received CE Mark approval in September 2007 and was also approved for sale by the Israeli Ministry of Health in 2008. We have maintained
our CE mark approval until now and expect that to continue going forward.
In
the European Union, UroShield has been marketed for the prevention of CAUTI and biofilm formation, decreased pain and discomfort associated
with urinary catheters and increased antibiotic efficacy.
The
company is continuing to assess the route for FDA clearance/approval.
UroShield
Sales and Marketing
From
time to time, we have had interest from strategic companies in the catheter market to partner, license or acquire the UroShield technology.
These strategic partners are active in the urology market and may be interested in integrating UroShield as an accessory, into their
respective range of products. Discussions with these partners are ongoing. There has also been interest from other companies with various
invasive line applications.
PainShield
PainShield
is an ultrasound device, consisting of a reusable driver unit and a disposable patch, which contains our proprietary therapeutic transducer.
It delivers a localized ultrasound effect to treat pain and induce soft tissue healing in a targeted area, while keeping the level of
ultrasound energy at a safe and consistent level of 0.4 watts.
We
believe the existing ultrasound therapy devices being used for pain reduction are primarily large devices used exclusively by clinicians
in medical settings. PainShield is able to deliver ultrasound therapy without being located in a health care facility or clinic because
it is portable, due to it being lightweight and battery operated. Because it is patch based and easy to apply, PainShield does not require
medical personnel to apply ultrasound therapy to the patient. Some patient benefits reported in prior studies included ease of application
and use, relatively quick recovery time, high patient compliance, and potentially increased safety and efficacy over certain other devices
that rely on higher-frequency ultrasound (Adahan M, et al, “A Sound Solution to Tendonitis: Healing Tendon Tears With a Novel Low-Intensity,
Low-Frequency Surface Acoustic Ultrasound Patch,” American Academy of Physical Medicine and Rehabilitation Vol. 2, 685-687, July
2010). PainShield can be used by patients at home or work or in a clinical setting and can be used even while the patient is sleeping.
Its range of applications includes acute and chronic pain reduction and anti-inflammatory treatment.
Picture
of PainShield with Patch
In
the United States, PainShield is only cleared to treat pain, muscle spasms, and joint contractures associated with or caused by various
conditions or diseases. It has also been used to treat pelvic and abdominal pain. To date, to the best of our knowledge, the primary
treatment options for several of these conditions are pain medication and surgery. Several additional causes of pain, and the treatment
of that pain with the PainShield product, can be explored through clinical trials.
Market
for PainShield
Pain-related
complaints are one of the most common reasons patients seek treatment from physicians (Prince V, “Pain Management in Patients with
Substance-Use Disorders,” Pain Management, PSAP-VII, Chronic Illnesses). According to Landro L, “New Ways to Treat Pain:
Tricking the Brain, Blocking the Nerves in Patients When all Else Has Failed,” Wall Street Journal, May 11, 2010, approximately
26% of adult Americans, or approximately 76.5 million people, suffer from chronic pain. The National Center for Health Statistics has
estimated that approximately 54% of the adult population experiences musculoskeletal pain. Studies have shown that low-frequency ultrasound
treatment has yielded positive results for a variety of indications, including tendon injuries and short-term pain relief (Warden SJ,
“A new direction for ultrasound therapy in sports medicine,” Sports Med. 2003; 33 (2):95-107), chronic low back pain (Ansari
NN, Ebadi S, Talebian S, Naghdi S, Mazaheri H, Olyaei G, Jalaie SA, “Randomized, Single Blind Placebo Controlled Clinical Trial
on the Effect of Continuous Ultrasound on Low Back Pain,” Electromyogr Clin Neurophysiol. 2006 Nov; 46(6):329-36) and sinusitis
(Ansari NN, Naghdi S, Farhadi M, Jalaie S, “A Preliminary Study Into the Effect of Low-Intensity Pulsed Ultrasound on Chronic Maxillary
and Frontal Sinusitis,” Physiother Theory Pract. 2007 Jul-Aug; 23(4):211-8). We believe that PainShield’s technology, portability
and ease of use may result in it becoming an attractive product in the pain management and therapy field.
Competition
for PainShield
There
are numerous products and approaches currently utilized to treat chronic pain. The pharmacological approach, which may be the most common,
focuses on drug-related treatments with the over-the-counter internal analgesic market estimated at $19 billion in 2019. Alternatively,
there are a large number of non-pharmacological pain treatment options available, such as ultrasound, transcutaneous electrical nerve
stimulation, or TENS, laser therapy and pulsed electromagnetic treatment. In addition, there are some technologies and devices in the
market that utilize low frequency ultrasound or patch technology. Many patients are initially prescribed anti-pain medication; however,
ongoing use of drugs may cause substantial side effects and lead to addiction. Therefore, patients and clinicians have shown increased
interest in alternative pain therapy using medical devices that do not carry these side effects.
The
currently available ultrasound treatments for chronic pain have generally been accepted by the medical community as standard treatment
for pain management. However, the traditional ultrasound treatments, such as those manufactured or distributed by Mettler Electronics
Corp, Metron USA and Zimmer MedizinSysteme, are stationary devices found only in clinics and other health care facilities that need to
be administered to patients by health care professionals. We are aware of three companies that market smaller ultrasound devices capable
of certain self-administered use for the treatment of pain: Koalaty Products, Inc., Sun-Rain System Corp. and PhysioTEC. These devices
generally function in the same manner, at the same frequency and with the same administration and safety requirements and limitations
as traditional, larger ultrasound devices. We are also aware of one product, the SAM® Sport4, which has recently received FDA approval
and also has CE Mark approval, marketed by ZetrOZ, Inc., that we understand may eliminate certain of these requirements and limitations,
namely the requirement to be plugged in, the need for movement around the treated area and the relatively short safe treatment period.
However, we understand that this product does not generate surface acoustic waves as our products do, which means that the treatment
area is generally limited to that under the transducer, that the use of transmission gel is still required, and that the transducer thickness
is significantly greater than ours (approximately 1.5cm). It is also our understanding that the FDA has issued contraindications which
do not apply to the PainShield product. In addition, there are other patch-based methods of pain treatment, such as TENS therapy. TENS
therapy may be painful and irritating for the patient due to the muscle contractions resulting from the electrical pulses.
PainShield
combines the efficacy of ultrasound treatment for pain with the ease of use and portability of a patch-based system. PainShield also
may be self-administered by the patient, including while the patient is sleeping. However, if we are unable to obtain widespread insurance
coverage and reimbursement for PainShield, its acceptance as a pain management treatment would likely be hindered, as patients may be
reluctant to pay for the product out-of-pocket.
CMS
approved PainShield for reimbursement for Medicare beneficiaries on a national basis in January 2020 although we have never received
a reimbursement value. The Company was denied reimbursement in September 2022 due to a lack of “life-cycle” testing. The
Company had engaged Carmel Labs in Israel to conduct this testing and submitted the results to CMS with our 2023 application on January
3, 2023. On August 21, 2023, CMS, denied reimbursement with respect to PainShield due to their request for additional longevity testing.
We are currently evaluating whether to resubmit another application to CMS.
Our
marketing efforts continue to expand in the direct to consumer, Veterans Health Care network, and workers’ compensation market.
Relative to the VA market, we are currently represented by Applied Medical and Delta Medical. Delta Medical is a Service Disabled Veteran
Organization Small Business (SDVOSB). PainShield is approaching the workers’ compensation market through various sales agents and
on a direct basis. Additionally, on March 1st, 2023, we established a rental program for direct to consumer marketing for
patients without health insurance coverage.
Regulatory
Strategy
PainShield
received 510(k) clearance from the FDA in August 2008 as an ultrasonic diathermy device intended to apply ultrasonic energy to generate
deep heat within body tissues for the treatment of selected medical conditions, such as relief of pain, muscle spasms, and joint contractures.
PainShield received CE Mark approval in July 2008.
In
the United States, a prescription from a licensed healthcare practitioner is required for the use of PainShield.
PainShield
Plus, also referred to as the PainShield MD Plus, is a dual applicator device that received FDA 510(k) clearance in November 2022. We
discontinued future sales of PainShield Plus following self-identification of certain inaccuracies in the 2022 510(k) application for
the PainShield Plus product. At the time we discontinued sales of PainShield Plus, we were unaware of any safety issue related to the
device, and we remain unaware of any such issues.
In
the United States, PainShield falls under the diathermy classification for the treatment of pain for initial reimbursement purposes.
The permitted reimbursement codes can be used in the outpatient supervised medical setting. We continue to work with the Centers for
Medicare and Medicaid Services and private insurers so that reimbursement can be extended to cover the administration of PainShield outside
of health care facilities and clinics. We have engaged outside legal counsel to assist with all aspects of reimbursement and FDA regulatory
actions. In addition, we intend to conduct clinical trials in order to pursue FDA authorization to market PainShield for a larger range
of indications. The targeted reimbursement would be based upon specific indications, where study data serves as justification for payment.
PainShield
Sales and Marketing
PainShield
was introduced in 2009 as a treatment for pain, such as tendonitis, sports injuries, pelvic pain, and neurologic pain, depending on
the scope of the approval or clearance from each applicable jurisdiction, and we have sold over 8,000 units since its introduction.
We currently maintain distribution agreements in the United States for the distribution of PainShield and continue to enhance our
marketing efforts both domestically and internationally to expand additional licensing and private label partnerships.
For
a discussion of the FDA approval process applicable to our products, as well as the regulation of our products generally, see “—Government Regulation” below.
Third
Party Reimbursement
We
anticipate that sales volumes and prices of the products we commercialize will depend in large part on the availability of coverage and
reimbursement from third party payers. Third party payers include governmental programs such as Medicare and Medicaid, private insurance
plans and workers’ compensation plans, Veterans Health Care network, among others. These third-party payers may deny coverage and
reimbursement for a product or therapy, in whole or in part, if they determine that the product or therapy was not medically appropriate
or necessary. The third-party payers also may place limitations on the types of physicians or clinicians that can perform specific types
of procedures. In addition, third party payers are increasingly challenging the prices charged for medical products and services. Some
third -party payers must also pre-approve coverage for new or innovative devices or therapies before they will reimburse health care
providers who use the products or therapies. Even though a new product may have been approved or cleared by the FDA for commercial distribution,
we may find limited demand for the device until adequate reimbursement has been obtained from governmental and private third -party payers.
In
international markets, reimbursement and healthcare payment systems vary significantly by country, and many countries have instituted
price ceilings on specific product lines and procedures. There can be no assurance that procedures using our products will be considered
medically reasonable and necessary for a specific indication, that our products will be considered cost-effective by third party payers,
that an adequate level of reimbursement will be available or that the third -party payers’ reimbursement policies will not adversely
affect our ability to sell our products profitably.
In
the United States, some insured individuals are receiving their medical care through managed care programs, which monitor and often require
pre-approval of the services that a member will receive. Some managed care programs are paying their providers on a per capita basis,
which puts the providers at financial risk for the services provided to their patients by paying these providers a predetermined payment
per member per month, and consequently, may limit the willingness of these providers to use certain products, including ours.
One
of the components in the reimbursement decision by most private insurers and governmental payers, including the Centers for Medicare
and Medicaid Services, which administers Medicare, is the assignment of a billing code. Billing codes are used to identify the procedures
performed when providers submit claims to third party payers for reimbursement for medical services. They also generally form the basis
for payment amounts.
Obtaining
reimbursement approval for a product from any government or other third -party payer is a time-consuming and costly process that could
require us or our distributors to provide supporting scientific, clinical and cost-effectiveness data for the use of our product to each
payer. Even if a code is obtained for a product, a third -party payer must still make coverage and payment determinations. When a payer
determines that a product is eligible for reimbursement, the payer may impose coverage limitations that preclude payment for some uses
that are approved by the FDA or other foreign regulatory authorities. We believe that the overall escalating costs of medical products
and services has led to, and will continue to lead to, increased pressures on the health care industry to reduce the costs of products
and services. In addition, health care reform measures, as well as legislative and regulatory initiatives at the federal and state levels,
create significant additional uncertainties. There can be no assurance that third party coverage and reimbursement will be available
or adequate, or that future legislation, regulation, or reimbursement policies of third -party payers will not adversely affect the demand
for our products or our ability to sell these products on a profitable basis. The unavailability or inadequacy of third -party payer
coverage or reimbursement would have a material adverse effect on our business, operating results and financial condition.
Effective as of January 2020, CMS approval for Medicare reimbursement was
added through Healthcare Common Procedure Coding System code K1004. We continue to work toward a favorable reimbursement rate with outside
legal counsel and reimbursement consultants.
Intellectual
Property
Intellectual
Property Related to Nano OpCo’s Business
Stemming
from a combination of patent, copyright, trademark and trade secret laws, as well as non-disclosure agreements and other contracts, our
intellectual property rights represent a vital resource to the management of our company. Therefore, we are continuing our practice of
investing in obtaining appropriate legal protection for our innovations whenever possible and have adopted a more fully integrative approach
to the management of our intellectual property that mutually aligns with our ongoing R&D strategies, commercial opportunities based
on market analyses, and longer-term business objectives.
From
our patented technologies to our trademarked brands, we believe our intellectual property has substantial value and has significantly
contributed to our success to date.
From
our patented technologies to our trademarked brands, we believe our intellectual property has substantial value and has significantly
contributed to our success to date.
We
seek patent protection for our inventions not only to differentiate our products and technologies, but also to develop opportunities
for licensing and securing our rights to profits therefrom. With the aim of optimizing commercial and regulatory success, our proprietary
technology and innovative applications thereof are protected by a variety of patent claims. We believe that our granted patents and pending
applications collectively protect our technology, both in terms of our existing products, as well as our anticipated pipeline of new
offerings.
Our
patent portfolio includes at least the following issued patents, as well as a number of corresponding foreign patents in relevant jurisdictions:
(1)
U.S. Patent No. 7,829,029 to “Acoustic Add-On Device for Biofilm Prevention in Urinary Catheter” (expiring on August 28,
2029). Foreign counterparts include: European Patent No. 1998834 B1, and Chinese Patent No. CN 101616707 B.
(2)
U.S. Patent No. 9,028,748 to “System and Method for Surface Acoustic Wave Treatment of Medical Devices” (expiring on July
11, 2030); and
(3)
U.S. Patent No. 9,585,977 directed to “System and Method for Surface Acoustic Waves Treatment of Skin” (expiring on August
20, 2033). Foreign counterparts include: European Patent No. EP 1991129 B1, Chinese Patent No. CN 101431940 B, and Israeli Patent No.
193600.
These
patents are directed to our proprietary surface acoustic wave (SAW) technology, including our commercialized PAINSHIELD, WOUNDSHIELD,
and UROSHIELD devices and the previously commercialized PAINSHIELD PLUS. Specifically, the patents provide for methods of generating
SAW on surfaces of indwelling medical devices and to topical and urological applications therefor, for alleviating pain and for wound
healing, and for preventing formation of bacterial biofilms on catheters.
In
addition to the above patents, our pending patent applications are representative of our ongoing efforts to broaden our portfolio as
we continue to develop new applications for our ultrasound technology. Pending patent applications related to UROSHIELD devices are
directed to Multiple Frequency Surface Acoustic Waves for Internal Medical Device (US Patent Application number 19/163,046)
and System, Device, and Method for Mitigating Bacterial Biofilms Associated with Indwelling Medical Devices,) US Patent
Application number 17/646,715, in restoration process). This patent application covers the next generation of UROSHIELD devices
operating at multiple frequencies and devices which are compatible in portable and wireless systems.
Pending
patent applications related to PAINSHIELD, PAINSHIELD PLUS, WOUNDSHIELD devices are directed to Transdermal Patch of a Portable
Ultrasound-Generating System for Improved Delivery of Therapeutic Agents and Associated Methods of Treatment (US Patent
Application no. 17/025,969) and Portable Ultrasound System and Methods of Treating Facial Skin by Application of Surface Acoustic
Waves) US Patent Application number 17/646,753, in restoration process.
Although
not yet granted, the aim of our growing number of patent applications is to secure our rights within additional industry sectors we foresee
as most readily benefiting from our technology. Therefore, looking beyond just pain management and urology, our patent applications relate
to, inter alia: novel transdermal patches uniquely configured to work with our ultrasound technology to additionally provide for
improved absorption and transdermal delivery of therapeutic agents during treatment; cosmetic applications of our ultrasound technology
to provide anti-aging benefits; and certain new or improved stand-alone therapeutic medical devices or so-called “indwelling medical
devices” (e.g., catheters, intravenous (IV) needle assemblies, and percutaneous endoscopic gastronomy (PEG) tubes) that include
our SAW-generating technology to provide the accompanying antimicrobial effect for preventing infections typically associated with available
indwelling devices.
We
intend to further grow our patent portfolio by continuing to patent new technology as it is developed, to defend intellectual property
as we believe necessary by actively pursuing any infringements, to pursue commercial opportunities our patents provide for our innovations,
and to continue to develop our brands and trademarks.
In
addition to patent protection, we own numerous registered trademarks for our commercialized WOUNDSHIELD (in the U.S. and Canada), NanoVibronix
(in the U.S. and Canada),
PAINSHIELD
(in the U.S. and Canada), and UROSHIELD (in the U.S.). Generally, the protection afforded by trademarks is perpetual, subject to paying
timely renewals and continuing proper use in commerce. In addition to the above, we expect to pursue additional trademark registrations
to the extent we believe they would be beneficial and cost-effective.
We
regularly enter into, and rely on, confidentiality and proprietary rights agreements with our employees, consultants, contractors and
business partners to protect our trade secrets, proprietary technology and other confidential information. We control the use of our
proprietary technology through relevant provisions, notifications, and disclaimers provided on our website, our customer terms of use,
and our vendor terms and conditions.
Intellectual
Property Related to ENvue Business
Below
is a brief overview of the status of ENvue’s main intellectual property assets as of February 6, 2025:
ENvue
regularly protects its intellectual property rights by filing patent applications in its main target market - the USA - as well as in
the main potential target markets for its future activities. Generally, the lifespan of these patents, is 20 years from the earliest
non-provisional patent filing date. These anticipated expiration dates are without taking into account any and all possible patent term
adjustments, extensions, or abandonments, and assuming payment of all appropriate maintenance, renewal, annuity, and other governmental
fees. ENvue continues to evaluate its intellectual property portfolio as patents reach end of life to determine the optimal course for
continuing to protect its technology. ENvue owns all its patents.
Below
are details about the significant registered patents and significant patent applications owned by ENvue:
| |
1. |
Nasogastric
Tube - A tube for insertion through the patient’s nose, intended for connection to a source of substances or pressure. |
| Country |
|
Status |
| Israel |
|
Granted |
| Germany |
|
Granted |
| United
States |
|
Granted
(5 patents) |
| China |
|
Granted
(2 patents) |
| |
2. |
Nasogastric
Tube - A tube for insertion through the patient’s nose, intended for connection to a source of substances or pressure.
The tube contains at least one main internal tube and one suction tube, which has at least one outlet used for suction with the purpose
of preventing damage to the patient’s internal tissues. |
| Country |
|
Status |
| Israel |
|
Granted |
| United
States |
|
Granted |
| |
3. |
Nasogastric
Tube - A system that includes a tube for insertion through the patient’s nose, containing a feeding mechanism, a suction
mechanism, and a gastric decompression mechanism. |
| Country |
|
Status |
| Europe
(Validated in AT, CH/LI, DE, ES, FR, GB, and IT) |
|
Granted |
| United
States |
|
Allowed |
| |
4. |
Enteral
Feeding Pump - A system of devices, including a pump for drawing fluids into the tube; a switching mechanism connected to at
least four internal tubes installed in the feeding tube; and a controller designed to operate the mentioned pump and switching mechanism. |
| Country |
|
Status |
| Israel |
|
Granted |
| United
States |
|
Granted |
| |
5. |
Insertion
Device Positioning Guidance System and Method - A system and method for guiding the insertion and positioning of a device within
a patient’s body. It includes an electromagnetic field generator that covers the treatment area, multiple sensors designed
to provide indications of the tube’s position within the patient’s digestive system and the patient’s posture.
Additionally, the system features a processor that collects and processes all data to create a three-dimensional anatomical map of
the patient’s upper body, all of which functions independently of patient movement and various deviations. |
| Country |
|
Status |
| China |
|
Granted |
| Japan |
|
Granted |
| United
States |
|
Granted
(5 patents) |
| |
6. |
Feeding
Tube with Electromagnetic Sensor - Feeding tubes that include an electromagnetic sensor and a wire that runs along the length
of the tube. |
| Country |
|
Status |
| Japan |
|
Granted |
| United
States |
|
Granted
(3 patents and 1 allowed application) |
| |
7. |
Insertion
Device Positioning Guidance System and Method - A system and method for guiding the insertion and positioning of a device within
a patient’s body, including an electromagnetic field generator that covers the treatment area, multiple sensors designed to
provide indications of the tube’s position within the patient’s digestive system, the patient’s posture, and other
relevant factors. The system also includes a processor that collects and processes all the data to align a predefined anatomical
map of a patient’s torso based on positions corresponding to locations on a patient’s upper body, all of which operates
independently of patient movement and other deviations. |
| Country |
|
Status |
| Europe
(Validated in AT, CH/LI, DE, ES, FR, GB, and IT) |
|
Granted |
| China |
|
Granted
(2 patents) |
| Japan |
|
Granted |
| United
States |
|
Granted
(3 patents) |
| |
8. |
Insertion
Device Positioning Guidance System and Method - A system and method for guiding the insertion and positioning of a device within
a patient’s body, which includes an electromagnetic field generator that covers the treatment area, multiple sensors designed
to provide indications of the tube’s position within the patient’s digestive system, the patient’s posture, and
other relevant factors. The system also includes a processor responsible for collecting and processing all the data to create a three-dimensional
anatomical map of the patient’s upper torso and to facilitate visualization on the anatomical map of a position, orientation
and/or path of a tip sensor, all of which functions independently of patient movement and other deviations. |
| Country |
|
Status |
| China |
|
2
Pending Applications |
| Japan |
|
Granted |
| United
States |
|
Granted
(4 patents) |
| |
9. |
Insertion
Device Positioning Guidance System and Method - A device, system, and method for guiding the insertion and positioning of an
insertion tube within the patient’s body based on sensing of changes in an electromagnetic field. |
| Country |
|
Status |
| Israel |
|
Pending |
| Japan |
|
Allowed |
| United
States |
|
Pending |
| |
10. |
Guidance
System with Claviculae Position Sensors - A device, system, and method for guiding the insertion and positioning of tube positioning
within the patient’s body based on sensing of changes in an electromagnetic field using sensors positioned on a patient’s
upper torso, where the calculation considers signals received from reference sensors located in the clavicle area of the patient. |
| Country |
|
Status |
| Israel |
|
Pending |
| Japan |
|
Allowed |
| United
States |
|
Pending |
As
of February 6, 2024, ENvue owns the following trademarks: Envizion Medical, ENsump, ENvue, ENgat, Envizion (wordmark and logo), and ENvue’s
logo in key countries, including the U.S., Europe, and China.
On
January 25, 2023, a request was submitted by Hologic, Inc.10 to narrow the list of goods described under the ENVIZION MEDICAL
trademark in the U.S. ENvue filed a partial voluntary surrender of its U.S. registration as to the following goods: Nasogastric aspiration
tube; Medical devices, namely, nasogastric tubes with integrated camera; Medical intubation equipment; nasogastric cameras for medical
purposes; Medical integrated camera for Nasogastric Aspiration Tubes; and Camera for placing a nasogastric tube in a patient’s
esophagus, which was accepted by the U.S. Patent and Trademark Office.
On
September 17, 2025, trademark applications for Oscar and the Oscar logo were filed in the U.S. and were approved
ENvue
also relies on trade secrets relating to its products and technology, including its data processing algorithms, and maintains the confidentiality
of such proprietary information to protect aspects of its business that are not amenable to, or that ENvue does not consider appropriate
for, patent protection. ENvue seeks to protect its trade secrets and know-how by entering into confidentiality and invention assignment
agreements with employees, contractors, consultants, suppliers, customers, and other third parties, who have access to such information.
These agreements generally provide that all confidential information concerning ENvue’s business or financial affairs developed
or made known to the individual during the course of the individual’s relationship with ENvue are to be kept confidential and not
disclosed to third parties except in specific circumstances. If any such person misappropriated ENvue’s trade secrets or other
know-how or confidential information, there is no guarantee that ENvue would be able to prevail in obtaining damages or injunctive relief
in a dispute regarding such misappropriation.
Despite
these protections, ENvue also notes that its employees may have been previously employed at other companies in the industry, including
its competitors or potential competitors. Although ENvue is not aware of any claims currently pending against it, ENvue may be subject
to claims that these employees or ENvue has inadvertently or otherwise used or disclosed trade secrets or other proprietary information
of the former employers of its employees. Litigation may be necessary to defend against these claims. Even if ENvue is successful in
defending against these claims, litigation could result in substantial costs and be a distraction to management. If ENvue fails in defending
such claims, in addition to paying money claims, ENvue may lose valuable intellectual property rights or personnel. A loss of key personnel
or their work product could hamper or prevent ENvue’s ability to commercialize product(s), which would materially adversely affect
its commercial development efforts.
As
of approximately February 6, 2025, ENvue holds design patents for Sump Tube in the U.S. and Tube Assembly for Feeding and Suction in
the U.S., Europe, the United Kingdom and China.
Government
Regulation
U.S.
Food and Drug Administration Regulation
Each
of our products must be approved, cleared by, or registered with the U.S. Food and Drug Administration (“FDA”) before they
can be marketed in the United States, and they can only be marketed consistently with their respective approved or cleared indication(s)
of use. Before and after approval or clearance in the United States, our products, approved or cleared products and product candidates,
are subject to extensive regulation by the FDA under the Federal Food, Drug, and Cosmetic Act and/or the Public Health Service Act, as
well as by other regulatory bodies. The FDA regulations govern, among other things, the development, testing, manufacturing, labelling,
safety, storage, record-keeping, market clearance or approval, advertising and promotion, import and export, marketing and sales, distribution
and market withdrawal and recalls of medical devices and pharmaceutical products.
FDA
Approval or Clearance of Medical Devices
In
the U.S., numerous laws and regulations govern the processes by which medical devices are developed, manufactured, brought to market
and marketed. These include the Federal Food, Drug, and Cosmetic Act (“FD&C Act”) and its implementing regulations issued
by FDA, among others. Unless an exemption applies, each medical device commercially distributed in the United States requires FDA clearance
of a 510(k) premarket notification (“510(k) clearance”), granting of a de novo request, or approval of an application for
premarket approval (“PMA”). In general, under the FD&C Act, medical devices are classified in one of three classes on
the basis of the controls necessary to reasonably assure their safety and effectiveness. A medical device’s classification determines
the level of FDA review and approval to which the device is subject before it can be marketed to consumers:
| |
● |
Class
I devices, the lowest-risk FDA device classification, include devices with the lowest risk to the patient and are those for which
safety and effectiveness can be assured by adherence to FDA’s medical device general controls, including labeling, establishment
registration, device product listing, adverse event reporting, and, for some products, adherence to good manufacturing practices
through FDA’s Quality System Regulations. |
| |
|
|
| |
● |
Class
II devices, moderate-risk devices, also require compliance with general controls and in some cases, special controls as deemed necessary
by FDA to ensure the safety and effectiveness of the device. These special controls may include performance standards, particular
labeling requirements, or post-market surveillance obligations. While most Class I devices are exempt from the 510(k) premarket notification
requirement, typically a Class II device also requires pre-market review and 510(k) clearance as well as adherence to the Quality
System Regulations/good manufacturing practices for devices. |
| |
|
|
| |
● |
Class
III devices, high-risk devices that are often implantable or life-sustaining or novel devices, also require compliance with the medical
device general controls and Quality System Regulations, and generally must be approved by FDA before entering the market through
a PMA application. Approved PMAs can include post-approval conditions and post-market surveillance requirements, analogous to some
of the special controls that may be imposed on Class II devices. |
PainShield
and the ENvue System are classified as Class II medical devices and require U.S. Food and Drug Administration authorization prior to
marketing, by means of 510(k) clearance. Due to its nature and the lack of existing predicate devices on the market, UroShield is automatically
classified as a Class III device for which a PMA is required, unless our request for De Novo reclassification is successful,
in which case, it will be classified as a Class II device and subject to the same post market framework as 510(k)-cleared devices.
To
request marketing authorization by means of a 510(k) clearance, we must submit a pre-market notification demonstrating that the proposed
device is substantially equivalent to a legally marketed medical device (referred to as a “predicate device”). A finding
of substantial equivalence requires that the proposed new device (i), has the same intended use as a predicate device; (ii) has the same
or similar technological characteristics as the predicate device; (iii) is as safe and effective as the predicate device; and (iv) does
not raise different questions of safety and effectiveness than the predicate device. 510(k) submissions generally include, among other
things, a description of the device and its manufacturing, device labelling, medical devices to which the device is substantially equivalent,
safety and biocompatibility information and the results of performance testing. In some cases, a 510(k) submission must include data
from human clinical studies. Marketing may commence only when the FDA issues a clearance letter finding substantial equivalence. The
typical duration to receive 510(k) approval is approximately nine months from the date of the initial 510(k) submission, although there
is no guarantee that the timing will not be longer.
The
FDA may require us to perform clinical studies to show a product candidate’s safety and efficacy in addition to technological equivalence
in support of our filed 510(k). No matter which regulatory pathway we may take in the future towards marketing products in the United
States, we believe we will be required to provide clinical proof of device effectiveness and safety.
After
a device receives 510(k) clearance, any product modification that could significantly affect the safety or effectiveness of the product,
or that would constitute a significant change in intended use, requires a new 510(k) clearance or, if the device would no longer be substantially
equivalent, would require a PMA. If the FDA determines that the product does not qualify for 510(k) clearance, then a company must submit
and the FDA must approve a PMA before marketing can begin. An alternative to a new 510(k) submission is a “letter to File”,
citing substantial equivalence to a product which has been granted 510(k) clearance.
A
PMA application must provide a demonstration of safety and effectiveness, which generally requires extensive nonclinical and clinical
trial data. Information about the device and its components, device design, manufacturing and labelling, among other information, must
also be included in the PMA. As part of the PMA review, the FDA will inspect the manufacturer’s facilities for compliance with
quality system regulation requirements, which govern testing, control, documentation and other aspects of quality assurance with respect
to manufacturing. If the FDA determines the application or manufacturing facilities are not acceptable, the FDA may outline the deficiencies
in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional
information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. During the review
period, an FDA advisory committee, typically a panel of clinicians and statisticians, is likely to be convened to review the application
and recommend to the FDA whether, or upon what conditions, the device should be approved. The FDA is not bound by the advisory panel
decision. While the FDA often follows the panel’s recommendation, there have been instances where the FDA has not. If the FDA finds
the information satisfactory, it will approve the PMA. The PMA approval can include post-approval conditions, including, among other
things, restrictions on labelling, promotion, sale and distribution, or requirements to do additional clinical studies post-approval.
Even after approval of a PMA, a new PMA or PMA supplement is required to authorize certain modifications to the device, its labelling
or its manufacturing process. Supplements to a PMA often require the submission of the same type of information required for an original
PMA, except that the supplement is generally limited to that information needed to support the proposed change from the product covered
by the original PMA. The typical duration to receive PMA approval is approximately two years from the date of submission of the initial
PMA application, although there is no guarantee that the timing will not be longer.
The
Food and Drug Administration Modernization Act of 1997 established a new route to market for low to moderate risk medical devices that
are automatically placed into Class III due to the absence of a predicate device, called the “Request for Evaluation of Automatic
Class III Designation,” or the De Novo classification procedure. This procedure allows a manufacturer whose novel device
is automatically classified into Class III to request down-classification of its medical device into Class I or Class II based on a benefit-risk
analysis demonstrating the device actually presents low or moderate risk, rather than requiring the submission and approval of a PMA
application. Prior to the enactment of the Food and Drug Administration Safety and Innovation Act of 2012, or FDASIA, a medical device
could only be eligible for De Novo classification if the manufacturer first submitted a 510(k) premarket notification and received
a determination from the FDA that the device was not substantially equivalent. FDASIA streamlined the De Novo classification pathway
by permitting manufacturers to request De Novo classification directly without first submitting a 510(k) premarket notification
to the FDA and receiving a not substantially equivalent determination. If the manufacturer seeks reclassification into Class II, the
manufacturer must include a draft proposal for special controls that are necessary to provide a reasonable assurance of the safety and
effectiveness of the medical device. In addition, the FDA may reject the reclassification petition if it identifies a legally marketed
predicate device that would be appropriate for a 510(k) or determines that the device is not low-to-moderate risk or that general controls
would be inadequate to control the risks and special controls cannot be developed. De Novo reclassification requests are also
subject to user fees, unless a specific exemption applies. If the device is not approved through De Novo review, then it must
go through the standard PMA process for Class III devices.
Clinical
Trials of Medical Devices
Clinical
trials are almost always required to support a PMA application and are sometimes required for a De Novo classification request
or 510(k) pre-market notification. In order to conduct a clinical investigation involving human subjects for the purpose of demonstrating
the safety and effectiveness of a medical device, an investigator acting on behalf of the company must, among other things, apply for
and obtain IRB approval of the proposed investigation. In addition, if the clinical study involves a “significant risk” (as
defined by the FDA) to human health, the company sponsoring the investigation must also submit and obtain FDA approval of an IDE. An
IDE must be supported by appropriate data, such as animal and laboratory testing results, showing that it is safe to test the device
in humans and that the testing protocol is scientifically sound. The IDE must be approved in advance by the FDA for a specified number
of study participants, unless the product is deemed a non-significant risk device and eligible for abbreviated IDE requirements. Generally,
clinical trials for a significant risk device may begin once the IDE is approved by the FDA and the study protocol and informed consent
are approved by a duly-appointed IRB at each clinical trial site.
FDA’s
IDE regulations govern investigational device labelling, prohibit promotion, and specify an array of GCP requirements, which include,
among other things, recordkeeping, reporting and monitoring responsibilities of study sponsors and study investigators. Clinical trials
must further comply with the FDA’s regulations for IRB approval and for informed consent and other human subject protections. Required
records and reports are subject to inspection by the FDA. The results of clinical testing may be unfavorable or, even if the intended
safety and efficacy success criteria are achieved, may not be considered sufficient for the FDA to grant approval or clearance of a product.
Post-Approval
Regulation of Medical Devices
After
a device is cleared or approved for marketing, numerous and pervasive regulatory requirements continue to apply. These include:
| |
● |
the
FDA quality systems regulation, which governs, among other things, how manufacturers design, test, manufacture, exercise quality
control over, and document manufacturing of their products; |
| |
|
|
| |
● |
labelling
and claims regulations, which prohibit the promotion of products for unapproved or “off-label” uses and impose other
restrictions on labelling; |
| |
|
|
| |
● |
if
applicable, the Electronic Product Regulations found in 21 CFR parts 1000-1050, which provide additional requirements applicable
to electronic products, including records and reporting requirements; and |
| |
|
|
| |
● |
the
Medical Device Reporting regulation, which requires reporting to the FDA of certain adverse experiences associated with use of the
product. |
Under
the FDA medical device reporting (“MDR”) regulations, medical device manufacturers are required to report to the FDA information
that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause
or contribute to death or serious injury if the malfunction of the device or a similar device of such manufacturer were to recur. The
decision to file an MDR involves a judgment by the manufacturer. If the FDA disagrees with the manufacturer’s determination, the
FDA can take enforcement action.
Additionally,
the FDA has the authority to require the recall of commercialized products in the event of material deficiencies or defects in design
or manufacture. The authority to require a recall must be based on an FDA finding that there is reasonable probability that the device
would cause serious adverse health consequences or death. Manufacturers may, under their own initiative, recall a product if any distributed
devices fail to meet established specifications, are otherwise misbranded or adulterated, or if any other material deficiency is found.
The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated.
The
failure to comply with applicable device regulatory requirements can result in enforcement action by the FDA, which may include any of
the following sanctions:
| ●
|
warning
letters, fines, injunctions, or civil penalties; |
| ● |
recalls,
detentions or seizures of products; |
| ●
|
operating
restrictions; |
| ● |
delays
in the introduction of products into the market; |
| ● |
total
or partial suspension of production; |
| ●
|
delay
or refusal of the FDA or other regulators to grant 510(k) clearance or PMA approvals of new products; |
| ● |
withdrawals
of marketing authorization; or |
| ●
|
in
the most serious cases, criminal prosecution. |
To
ensure compliance with regulatory requirements, medical device manufacturers are subject to market surveillance and periodic, pre-scheduled
and unannounced inspections by the FDA, and these inspections may include the manufacturing facilities of subcontractors and third-party
component suppliers.
Good
Manufacturing Practices Requirements
As
noted above, manufacturers of medical devices are required to comply with the good manufacturing practices set forth in the quality system
regulations promulgated under section 520 of the Food, Drug and Cosmetic Act as further set forth in the Code of Federal Regulations
as 21 CFR Part 820. Current good manufacturing practices (“CGMP”) regulations require, among other things, quality control
and quality assurance as well as the corresponding maintenance of records and documentation. The manufacturing facility for an approved
product must meet current good manufacturing practices requirements to the satisfaction of the FDA pursuant to a pre-PMA approval inspection
before the facility can be used. Manufacturers, including third party contract manufacturers, are also subject to periodic inspections
by the FDA and other authorities to assess compliance with applicable regulations. Failure to comply with or to promptly comply with
statutory and regulatory requirements subjects a manufacturer, and possibly us, to possible legal or regulatory action, including the
seizure or recall of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations,
and civil and criminal penalties. Adverse experiences with the product must be reported to the FDA and could result in the imposition
of marketing restrictions through labelling changes or in product recall. Product approvals may be withdrawn if compliance with regulatory
requirements is not maintained or if problems concerning safety or efficacy of the product occur following the approval.
International
Regulation
We
are subject to regulations and product registration requirements in many foreign countries in which we may sell our products, including
in the areas of product standards, packaging requirements, labelling requirements, import and export restrictions and tariff regulations,
duties and tax requirements. The time required to obtain clearance required by foreign countries may be longer or shorter than that required
for FDA clearance, and requirements for licensing a product in a foreign country may differ significantly from UFDA requirements.
There
is currently no premarket government review of medical devices in the European Economic Area (“EEA”). However, all medical
devices placed on the market in the EEA must meet the relevant essential requirements laid down in Annex I of Directive 93/42/EEC concerning
medical devices, or the Medical Devices Directive. The most fundamental essential requirement is that a medical device must be designed
and manufactured in such a way that it will not compromise the clinical condition or safety of patients, or the safety and health of
users and others. In addition, the device must achieve the performances intended by the manufacturer and be designed, manufactured, and
packaged in a suitable manner. The European Commission has adopted various standards applicable to medical devices. These include standards
governing common requirements, such as sterilization and safety of medical electrical equipment, and product standards for certain types
of medical devices. There are also harmonized standards relating to design and manufacture. While not mandatory, compliance with these
standards is viewed as the easiest way to satisfy the essential requirements as a practical matter. Compliance with a standard developed
to implement an essential requirement also creates a rebuttable presumption that the device satisfies that essential requirement.
In
the European Union, the European Medicines Agency and the European Union Commission determined that PainShield, UroShield, and WoundShield
are to be regulated as medical device products. These products are classified as Class II devices. These devices are CE Marked and as
such can be marketed and distributed within the European Economic Area. We are required to be recertified each year for CE by Intertek,
which conducts an annual audit. The ENvue System received a European CE mark, indicating that ENvue affirms its product’s conformity
with European health, safety and environmental protection standards, in 2021.The audit procedure, which includes on-site visits at our
facility, requires us to provide Intertek with information and documentation concerning our management system and all applicable documents,
policies, procedures, manuals, and other information.
On
April 5, 2017, the European Parliament passed the Medical Devices Regulation (Regulation 2017/745), which repeals and replaces the EU
Medical Device Directive and became effective on May 26, 2021. The Medical Devices Regulation, among other things, is intended to establish
a uniform, transparent, predictable, and sustainable regulatory framework across the EEA for medical devices and ensure a high level
of safety and health while supporting innovation. The new regulations, among other things:
| |
● |
strengthen
the rules on placing devices on the market and reinforce surveillance once they are available; |
| |
● |
improve
the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| |
● |
set
up a central database to provide patients, healthcare professionals, and the public with comprehensive information on products available
in the E.U.; and |
| |
● |
strengthen
rules for the assessment of certain high-risk devices, such as implants, which may have to undergo an additional check by experts
before they are placed on the market. |
The
primary regulatory bodies and paths in Asia, Australia, and Latin America are determined by the requisite country authority. In most
cases, establishment registration and device licensing are applied for at the applicable Ministry of Health through a local intermediary.
European
Good Manufacturing Practices
In
the European Union, the manufacture of medical devices is subject to good manufacturing practice, as set forth in the relevant laws and
guidelines of the European Union and its member states. Compliance with good manufacturing practice is generally assessed by the competent
regulatory authorities. Typically, quality system evaluation is performed by a notified body, which also recommends to the relevant competent
authority for the European Community CE Marking of a device. The competent authority may conduct inspections of relevant facilities,
and review manufacturing procedures, operating systems and personnel qualifications. In addition to obtaining approval for each product,
in many cases each device manufacturing facility must be audited on a periodic basis by the notified body. Further inspections may occur
over the life of the product.
U.S.
Fraud and Abuse and Other Health Care Laws
In
the United States, federal and state fraud and abuse laws prohibit the payment or receipt of kickbacks, bribes or other remuneration
intended to induce the purchase or recommendation of health care products and services. Other provisions of federal and state laws prohibit
presenting, or causing to be presented, to third party payers for reimbursement, claims that are false or fraudulent, or which are for
items or services that were not provided as claimed. In addition, other health care laws and regulations may apply, such as transparency
and reporting requirements, and privacy and security requirements. Violations of these laws can lead to civil and criminal penalties,
including exclusion from participation in federal and state health care programs. These laws are potentially applicable to manufacturers
of products regulated by the FDA as medical devices, such as us, and hospitals, physicians and other potential purchasers of such products.
The health care laws that may be applicable to our business or operations include:
| |
● |
Federal
false claims laws and civil monetary penalty laws, including the False Claims Act, that prohibit,
among other things, individuals or entities from knowingly presenting, or causing to be presented,
claims for payment from Medicare, Medicaid or other government health care programs that
are false or fraudulent, or making a false statement to avoid, decrease or conceal an obligation
to pay money to the federal government.
|
|
● |
The
federal Anti-Kickback Statute, which prohibits the offer, payment, solicitation or receipt
of any form of remuneration in return for referring, ordering, leasing, purchasing or arranging
for, or recommending the ordering, purchasing or leasing of, items or services payable by
Medicare, Medicaid or any other federal health care program
|
| |
● |
The
federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which prohibits knowingly and willfully executing,
or attempting to execute, a scheme to defraud any healthcare benefit program or obtain, by means of false or fraudulent pretenses,
representations, or promises, any of the money or property owned by, or under the custody or control of, any health care benefit
program, and for knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false statements
in connection with the delivery of or payment for health care benefits, items or services. |
| |
|
|
| |
● |
HIPAA,
as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, and its implementing regulations, which
also impose obligations and requirements on health care providers, health plans, and healthcare clearinghouses as well as their respective
business associates that perform certain services for them that involve the use or disclosure of individually identifiable health
information, with respect to safeguarding the privacy and security of certain individually identifiable health information. |
| |
|
|
| |
● |
The
federal transparency requirements under the Affordable Care Act, including the provision commonly referred to as the Physician Payments
Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies that are reimbursable under
Medicare, Medicaid or Children’s Health Insurance Program to report annually to Centers for Medicare and Medicaid Services,
or CMS, information related to payments and other transfers of value to physicians and teaching hospitals, and ownership and investment
interests held by physicians and their immediate family members. |
| |
|
|
| |
● |
Analogous
state and foreign laws and regulations, such as state anti-kickback and false claims laws, which may be broader in scope and apply
to referrals and items or services reimbursed by both governmental and non-governmental third-party payers, including private insurers,
many of which differ from each other in significant ways and often are not pre-empted by federal law, thus complicating compliance
efforts. |
Health
Information Privacy and Security Laws
There
are numerous U.S. federal and state laws and regulations related to the privacy and security of Personally Identifiable Information (“PII”),
including health information. Among others, the federal Health Insurance Portability and Accountability Act of 1996, as amended by HITECH,
and their implementing regulations, which we collectively refer to as HIPAA, establish privacy and security standards that limit the
use and disclosure of Protected Health Information (“PHI”) and require covered entities and business associates to implement
administrative, physical, and technical safeguards to ensure the confidentiality, integrity, and availability of individually identifiable
health information in electronic form, among other requirements.
Violations
of HIPAA may result in civil and criminal penalties. Our hospital customers are typically covered entities under HIPAA, and we are therefore
limited in the health information we may collect, receive, use, and disclose. To the extent we provide services that require the use
of PHI, we may be business associates of such covered entities and directly subject to HIPAA.
State
attorneys general also have the right to prosecute HIPAA violations committed against residents of their states, and HIPAA standards
have been used as the basis for the duty of care in state civil suits, such as those for negligence or recklessness in misusing personal
information. In addition, HIPAA mandates that HHS conduct periodic compliance audits of HIPAA covered entities and their business associates
for compliance.
Many
states also have laws that protect the privacy and security of sensitive and personal information, including health information. These
laws may be similar to or even more protective than HIPAA and other federal privacy laws. For example, the laws of the State of California
are more restrictive than HIPAA. Where state laws are more protective than HIPAA, we must comply with the state laws we are subject to.
California passed the California Consumer Privacy Act or CCPA on June 28, 2018, which went into effect January 1, 2020. On November 3,
2020, the California Privacy Rights Act of 2020 (“CPRA”), which amends the CCPA and adds new privacy protections that became
effective on January 1, 2023, was enacted through a ballot initiative. Records and information we maintain on our patients may be subject
to the CCPA if it is not covered by HIPAA. In certain cases, it may be necessary to modify our planned operations and procedures to comply
with these more stringent state laws. Not only may some of these state laws impose fines and penalties upon violators, but also some,
unlike HIPAA, may afford private rights of action to individuals who believe their personal information has been misused. In addition,
state and federal privacy laws are subject to frequent change.
In
addition to HIPAA and state health information privacy laws, we may be subject to or restricted by other state and federal privacy laws,
including laws that prohibit unfair privacy and security practices and deceptive statements about privacy and security, laws that place
specific requirements on certain types of activities, such as data security and texting, and laws requiring holders of personal information
to maintain safeguards and to take certain actions in response to a data breach.
Foreign
data protection, privacy, and other laws and regulations are often more restrictive than those in the U.S. The E.U., for example, traditionally
has imposed stricter obligations under its laws and regulations relating to privacy, data protection and consumer protection than the
U.S. In May 2018, the GDPR, governing data practices and privacy in the E.U., became effective and replaced the data protection laws
of the individual member states. GDPR requires companies to meet stringent requirements regarding the handling of personal data of individuals
in the E.U. These more stringent requirements include expanded disclosures to inform members about how we may use their personal data,
increased controls on profiling members, and increased rights for members to access, control and delete their personal data. In addition,
there are mandatory data breach notification requirements. The law also includes significant penalties for non-compliance, which may
result in monetary penalties of up to 20 million Euros or 4% of a company’s worldwide turnover, whichever is higher. GDPR and other
similar regulations require companies to give specific types of notice and informed consent is required for the placement of a cookie
or similar technologies on a user’s device for online tracking for behavioral advertising and other purposes and for direct electronic
marketing, and the GDPR also imposes additional conditions in order to satisfy such consent, such as a prohibition on pre-checked consents.
It remains unclear how the U.K. data protection laws or regulations will develop in the medium to longer term and how data transfer to
the U.K. from the E.U. will be regulated. Outside of the E.U., there are many other countries with data protection laws, and new countries
are adopting data protection legislation with increasing frequency.
Many
of these laws may require consent from individuals for the use of data for various purposes, including marketing, which may reduce our
ability to market our products.
There
is no harmonized approach to these laws and regulations globally. Consequently, we increase our risk of non-compliance with applicable
foreign data protection laws and regulations when we expand internationally. We may need to change and limit the way we use personal
information in operating our business and may have difficulty maintaining a single operating model that is compliant. Compliance with
such laws and regulations will result in additional costs and may necessitate changes to our business practices and divergent operating
models, limit the effectiveness of our marketing activities, adversely affect our business, results of operations, and financial condition,
and subject us to additional liabilities.
Manufacturing
and Suppliers
ENvue
Products
ENvue
does not manufacture the products it sells. ENvue has contracted with suppliers for the supply of raw materials and various components
that make up the system, as well as for the assembly of the system and the specialized feeding tubes.
We
estimate that there may be a limitation on the potential annual production capacity at the supplier that assembles the ENvue System.
ENvue’s management previously estimated that it can contract with an additional manufacturer if necessary. It should also be noted
that several companies with a global presence provide similar services to those of the current manufacturer, and ENvue previously estimated
that, if necessary, it could replace or expand the existing assembly capabilities within 6-9 months without significant cost changes.
customers.
Additionally,
there are only a few manufacturers worldwide that produce feeding tubes (made of polyurethane) used by ENvue to produce the specialized
feeding tubes for the ENvue System. Accordingly, terminating the contract with our feeding tube supplier could affect the production
capacity for the ENvue System for the time required to reorganize until we contract with an alternative supplier.
Nano
OpCo’s Products
For
the year 2025, through the current date, all of our programming and disposable kit manufacturing are being performed in our facilities
in Israel.
We
order certain component parts on an as-needed basis, generally from the manufacturer that provides us with the most competitive pricing.
Our most significant suppliers for these components are B Star, Inc and Plastic One. We do not have written agreements with any of these
suppliers, but we believe anyone could be replaced if necessary.
Customers
We
currently sell our products both directly through our website and indirectly via distribution agreements, with approximately 98% of our
sales coming through distributors and customers who are referred to us through sales agents, and the remaining 2% from consumers who
contact us through our website. We have exclusive and non-exclusive distribution agreements for our products with medical product distributors
based in the United States, in the United Kingdom and various countries throughout Europe, Australia, New Zealand, and Malta. For the
year ended December 31, 2025, our largest customer was Ultra Pain Products Inc, to whom our sales of products to them comprised approximately
31% of our total revenues.
We
are currently in discussions with several distribution companies with access to various markets in the United States, Europe, and Asia,
as well as the Veterans Health Care network facilities. Our current agreements stipulate that distributors will be responsible for carrying
out local marketing activities and sales. In addition, in most cases, all sales costs, including sales representatives, incentive programs, and marketing trials, will
be borne by the distributor. We expect any future distribution agreements to contain substantially similar stipulations. Under our current
agreements, distributors purchase our products from us at a fixed price. Our current agreements with distributors are generally for a
term of approximately two to three years and automatically renew for additional annual terms unless modified by either party. We also
service patients directly as a result of independent sales agents.
Our
People and Human Capital Resources
Employees
As
of December 31, 2025, ENvue has 12 full-time employees and 2 part-time employees. We also regularly work with several independent
consultants and other contract organizations to support our business and we regularly evaluate additional talent to help support our
product manufacturing, development, financial, and other capabilities.
Diversity
and Inclusion
We
believe that an inclusive culture is required to understand and develop products that benefit all patients. By embracing differences,
we aim to foster an environment of respect and trust in an effort to facilitate creativity, spark passion, and help us achieve better
outcomes for all those who work at the Company. We are committed to creating and maintaining a workplace free from discrimination or
harassment, including on the basis of any class protected by applicable law, and our recruitment, hiring, development, training, compensation,
and advancement practices are based on qualifications, performance, skills, and experience without regard to gender, race, or ethnicity.
Our management team and employees are expected to exhibit and promote honest, ethical, and respectful conduct in the workplace, including
adhering to the standards for appropriate behavior set forth in our code of conduct.
Compensation
and Benefits
We
operate in a highly competitive environment for human capital, particularly as we seek to attract and retain talent with relevant experience
in the medical device sector. Therefore, we strive to provide a total rewards package to our employees that is competitive with our peer
companies, including competitive healthcare benefits and in certain cases, stock options. We also offer paid leave as mandated by government
regulations, flexible work schedules, and other benefits as mandated by government regulations.
We
also offer employees the benefit of equity ownership in ENvue through stock option grants. We believe these grants both help promote
alignment between our employees and our stockholders and provide retention benefits, as the awards generally vest over a three-year period.
We
do not have any employees that are represented by a labor union or that have entered into a collective bargaining agreement with the
Company.
Safety
and Wellness
At
ENvue, we believe that health matters to everyone, and the safety health, and wellness of our employees is one of our top priorities.
We are committed to developing and fostering a work environment that is safe, professional, and promotes teamwork, diversity, and trust
in order to afford all of our employees the opportunity to contribute to the best of their abilities.
Seasonality
The
Company’s field of activity is not characterized by seasonality. It should be noted that hospitals, which are the current and potential
customers of the ENvue System, tend to concentrate their purchases of medical equipment in the last quarter of the year (end of the fiscal
year).
Legal
Proceedings
From
time to time, we may become party to legal proceedings in the ordinary course of business. Such legal proceedings may negatively impact
our business and financial position, result in brand or reputational harm, and divert the attention of our management from core operations
of our business.
Protrade
Proceeding
On
February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the
International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach
of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges,
in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of
an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million. The Company vigorously defended the claims
asserted by Protrade.
On
March 15, 2022, the arbitrator issued a final award, which, determined that (i) the Company had the right to terminate the Exclusive
Distribution Agreement; (ii) the Company did not breach the duty of good faith and fair dealing with regard to the Exclusive Distribution
Agreement; and (iii) the Company did not breach any confidentiality obligations to Protrade. Nevertheless, the arbitrator determined
that the Company did not comply with the obligation to supply Protrade with a year’s supply of patches, and awarded Protrade $1,500,250,
which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the
Company allegedly failed to supply Protrade with certain patches utilized by users of DV0057 Painshield MD device. The arbitrator based
the decision on the testimony of Protrade’s president who asserted that a user would use in excess of 33 patches per each device.
The Company believes that the number of patches per device alleged by Protrade is grossly inflated, and that these claims were not properly
raised before the arbitrator. Accordingly, on April 13, 2022, the Company submitted an application for the correction of the award which
the arbitrator denied on June 22, 2022.
On
April 5, 2022, Protrade filed a Petition with the Supreme Court of New York Nassau County seeking to confirm the Award. On April 13,
2022, the Company submitted an application to the ICA seeking to correct an error in the award based on the evidence that the Company
only sold 2-3 reusable patches per device contrary to the 33 reusable patches claimed by Protrade. The same arbitrator who issued the
award, denied the application.
On
July 22, 2022, the Company filed a cross-motion seeking to vacate arbitration award on the grounds that the arbitrator exceeded her authority,
that the award was procured by fraud, and that the arbitrator failed to follow procedures established by New York law. In particular,
the Company averred in its motion that Protrade’s witness made false statements in arbitration, and that the arbitrator resolved
a claim that was never raised by Protrade and that has no factual basis.
On
October 3, 2022, the court issued a decision granting Protrade its petition to confirm the award and denying the cross-motion.
On
November 9, 2022, the Company filed a motion to re-argue and renew its cross-motion to vacate the arbitration decision based on newer
information that was not available during the initial hearing. On the same day, the Company also filed a notice of appeal with the Appellate
Division, Second Department. On March 21, 2023, the court denied the motion to re-argue and renew.
On
July 10, 2023, the Company filed its appeal with the Appellate Division, Second Department. That
appeal is now fully briefed. In February 2025, the Second Department informed counsel for the Company that the Second Department was
beginning to process the appeal for calendaring.
On
March 30, 2026, the Appellate Division of the Second Department issued a decision and order which affirmed the judgment of the Supreme
Court, Nassau County in its entirety and dismissed the appeal, stating inter alia that “NanoVibronix failed to establish that the
arbitration award violated a strong public policy, was irrational, procured by fraud, or clearly exceeded a specifically enumerated limitation
of the arbitrator’s power”. It further held that NanoVibronix had not demonstrated that it had shown sufficient new facts
to the court on the motion to renew. The decision was conclusory and did not analyze the facts as argued by NanoVibronix nor did it distinguish
them specifically. The Company is currently reviewing the decision and considering its available alternatives.
As
of December 31, 2025, and 2024, the Company accrued the amount of the arbitration award to Protrade of approximately $2.3 million and
$2.1 million, respectively, including interest which is classified in “Other accounts payable and accrued expenses”.
Initiation
of Insolvency Proceedings Against Predecessor ENvue
During
2023, Predecessor ENvue was involved in the development of components and products to improve and upgrade the system as part of its research
and development activities, in order to optimize its use, expand its advantages and capabilities, and increase the potential target market.
As part of efficiency processes and cost-cutting measures in Predecessor ENvue, as of July 2023, Predecessor ENvue ceased its research
and development activities. The activities were resumed following the purchase by ENvue Holdings Corp.
Available
Information
The
Company’s Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and amendments thereto, are
filed with the Securities and Exchange Commission (the “SEC”). The Company is subject to the informational requirements of
the Securities Exchange Act of 1934, as amended (the “Exchange Act”) and files or furnishes reports, proxy statements and
other information with the SEC. Such reports and other information filed by the Company with the SEC are available free of charge on
the Company’s website at www.envuemed.com, as soon as reasonably practicable after we have electronically filed with, or
furnished to, the SEC. The SEC maintains an internet site that contains reports, proxy and information statements and other information
regarding issuers that file electronically with the SEC at www.sec.gov. The contents of these websites are not incorporated into
this filing. Further, the Company’s references to website URLs are intended to be inactive textual references only.
ITEM
1A. RISK FACTORS
Investing
in our common stock involves a high degree of risk. Before investing in our securities, you should carefully consider the following risks,
together with the financial and other information contained in this Annual Report on Form 10–K for the year ended December 31,
2025, and our other periodic filings with the SEC. Additional risks and uncertainties that we are unaware of may become important factors
that affect us. If any of the following events occur, our business, financial conditions and operating results may be materially and
adversely affected. In that event, the trading price of our common stock may decline, and you could lose all or part of your investment.
Summary
of Risk Factors
Below
is a summary of the principal factors that make an investment in our common stock speculative or risky. This summary does not address
all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face,
can be found below under the heading “Risk Factors” and should be carefully considered, together with other information in
this Form 10-K and our other filings with the SEC, before making an investment decision regarding our securities.
Risks
Related to NanoVibronix’s Business
| |
● |
We
have a history of losses, and we expect to continue to incur losses and may not achieve or maintain profitability. |
| |
● |
If
we are unable to raise additional capital, our clinical trials and product development will be limited and our long-term viability
will be threatened; however, if we do raise additional capital, your percentage ownership as a stockholder could decrease and constraints
could be placed on the operations of our business. |
| |
● |
If
we fail to obtain an adequate level of reimbursement for our approved products by third party payers, there may be no commercially
viable markets for our approved products or the markets may be much smaller than expected. |
| |
● |
We
face the risk of product liability claims and may not be able to obtain insurance. |
| |
● |
Our
product candidates may not be developed or commercialized successfully. |
| |
● |
Our
need to increase the size of our organization in order to successfully manage our growth. |
| |
● |
Our
failure to protect our intellectual property rights could diminish the value of our solutions, weaken our competitive position and
reduce our revenue. |
| |
● |
Our
financial statements have been prepared on a going concern basis, and do not include adjustments that might be necessary if we are
unable to continue as a going concern. Management has substantial doubt about our ability to continue as a going concern. |
| |
|
Our
business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security. |
Risks
Related to the Regulation of NanoVibronix’s Products
| |
● |
We
are subject to extensive governmental regulation, including the requirement of U.S. Food and Drug Administration approval or clearance
before our product candidates may be marketed and after approval or clearance and during the marketing of our products. |
| |
● |
UroShield
has not been cleared or approved by the FDA, nor has it undergone the same type of review as an FDA-approved or cleared device. |
| |
● |
Failure
to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products abroad. |
| |
● |
We
are uncertain regarding the success of our clinical trials for our products in development. |
| |
● |
Healthcare
reform measures could adversely affect our business and financial results. |
Risks
Related to NanoVibronix’s Operations in Israel
| |
● |
We
conduct our operations in Israel and therefore our results may be adversely affected by political, economic and military instability
in Israel and its region. |
| |
● |
Because
a certain portion of our expenses is incurred in currencies other than the U.S. dollar, our results of operations may be harmed by
currency fluctuations and inflation. |
Risks
Related to ENvue’s Organization and Securities
| |
● |
The
price of our securities may be volatile, and the market price of our securities may drop below the price you pay. |
| |
● |
We
have a significant number of warrants and options, and future sales of our common stock upon exercise of these options or warrants,
or the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is
doing well. |
| |
● |
Although
our shares of common stock are listed on Nasdaq, we currently have a limited trading volume, which results in higher price volatility
for, and reduced liquidity of, our common stock. |
| |
● |
If
we fail to comply with the continued listing requirements of Nasdaq, our common stock may be delisted and the price of our common
stock and our ability to access the capital markets could be negatively impacted. |
| |
● |
If
we fail to maintain effective internal control over financial reporting, our business, financial condition or results of operations
may be adversely affected. |
Risks
Related to ENvue’s Financial Condition, Business and Operations
| |
● |
The
financial statement footnotes of the Company include disclosure regarding the substantial doubt about the ability of the Company
to continue as a going concern. |
| |
● |
We
conduct certain of our operations in Israel. Conditions in Israel, including the October 2023 attack by Hamas and other terrorist
organizations from the Gaza Strip and Israel’s war against them, may affect our operations. |
| |
● |
The
impact of planned changes in the Israeli Judicial System on capital raising in the high-tech sector is difficult to predict. |
Risks
Related to the ENvue System
| |
● |
If
we are not successful in enhancing awareness of our ENvue System, driving adoption across our current target population and expanding
the population of eligible patients, our sales, business, financial condition and results of operations will be negatively affected. |
| |
● |
Our
commercial success and revenues will depend on the future adoption of the ENvue System into patient work streams in facilities and
other healthcare settings. If we are unable to successfully drive interest in our ENvue System, our business, financial condition
and results of operations would be harmed. |
| |
● |
We
may be unable to compete successfully with competitive technologies, which could harm our sales, business, financial condition and
results of operations. |
| |
● |
Use
of our ENvue System requires appropriate training and inadequate training may lead to negative clinician experiences, which could
harm our business, financial condition, and results of operations. |
| |
● |
Future
sales of our ENvue System may depend on providers’ and patients’ ability to obtain reimbursement from third-party payors,
such as insurance carriers. |
Risks
Related to ENvue Legal, Regulatory and Compliance Matters
| |
● |
Complying
with regulations enforced by the FDA and other regulatory authorities is expensive and time consuming, and failure to comply could
result in substantial penalties. |
| |
● |
We
may not receive the necessary authorizations to market future versions, if any, of our ENvue System or any future new product candidates,
and any failure to timely do so may adversely affect our ability to grow our business. |
| |
● |
Certain
modifications to our products may require new 510(k) clearance or other marketing authorizations. |
Risks
Related to ENvue’s Intellectual Property
| |
● |
Our
success depends in part on our proprietary technology, and if we are unable to successfully enforce our intellectual property rights,
our competitive position may be harmed. |
| |
● |
If
we infringe or violate the patents or proprietary rights of other parties or are subject
to an intellectual property infringement or misappropriation claim, our ability to grow our
business may be severely limited.
|
Risks
Related to Our Business
We
have a history of recurring losses and our financial statements have been prepared on a going concern basis, and do not include adjustments
that might be necessary if we are unable to continue as a going concern. If we are unable to raise capital when needed, we could be forced
to delay, reduce or eliminate any product development programs or commercialization efforts.
For
the fiscal year ended December 31, 2025, and 2024, we had a net loss of approximately $18.2 million and $3.7 million, respectively, with
revenues of approximately $2.6 million and $2.6 million, respectively. As of December 31, 2025, and 2024, we had an accumulated deficit
of approximately $90.5 million and $69.8 million, respectively. We expect to incur losses for at least the next year, as we continue
to incur expenses related to seeking additional U.S. Food and Drug Administration (“FDA”) clearances for PainShield, and
market acceptance of PainShield, which may require costly additional clinical trials and research, further product development and professional
fees associated with regulatory compliance.
Our
unaudited condensed consolidated financial statements have been prepared on a going concern basis, which contemplates the realization
of assets and the satisfaction of liabilities in the normal course of business. During the year ended December 31, 2025, our cash used
in operations was $9.4 million leaving a cash balance of $4.2 million as of December 31, 2025. Because we do not have sufficient resources
to fund our operations for the next twelve months from the date of this filing, management has substantial doubt about our ability to
continue as a going concern. In addition, we have incurred additional short-term debt related to the Merger. The consolidated financial
statements do not include any adjustments relating to the recoverability and classification of asset amounts or the classification of
liabilities that might be necessary should we be unable to continue as a going concern.
We
will need to raise additional capital to finance our losses, debt obligations, and negative cash flows from operations and may continue
to be dependent on additional capital raising as long as our products do not reach commercial profitability. There are no assurances
that we would be able to raise additional capital on terms favorable to it. If we are unsuccessful in commercializing our products and
raising capital, we will need to reduce activities, curtail, or cease operations. Even if we succeed in commercializing our new products,
we may not be able to generate sufficient revenues to cover our expenses and achieve profitability or be able to maintain profitability.
The
intended benefits of the Merger may not be realized.
The
Merger poses risks for our ongoing operations, including, among others:
| ● |
changes
in the business operations, strategies and focus of the combined company following the Merger may not result in an improvement in
the value of our common stock.; |
| |
|
| ● |
costs
and expenses associated with any undisclosed or potential liabilities; and |
| |
|
| ● |
unforeseen
difficulties may arise in integrating ENvue Holdings’ business in the combined company. |
Our
NanoVibronix business differs from that of ENvue Holdings in important respects and, accordingly, the results of operations of the combined
company and the market price of our common stock following the Merger may be affected by factors different from those affecting our results
of operations prior to the Merger. Market prices for securities of life sciences and medical technology companies in particular have
historically been particularly volatile and have shown extreme price and volume fluctuations that have often been unrelated or disproportionate
to the operating performance of those companies. Broad market and industry factors, as well as general economic, political and market
conditions such as recessions or interest rate changes, may seriously affect the market price of the combined company’s common
stock, regardless of our actual operating performance. See “Risks Related to Our Organization and Securities - The price of our
securities may be volatile, and the market price of our securities may drop below the price you pay.”
In
the past, following periods of volatility in the overall market and the market prices of particular companies’ securities, securities
class action litigation has often been instituted against these companies. Litigation of this type, if instituted against us, could result
in substantial costs and a diversion of management’s attention and resources. Any adverse determination in any such litigation
or any amounts paid to settle any such actual or threatened litigation could require that the combined company make significant payments.
As
a result of the foregoing, we may be unable to realize the full strategic and financial benefits previously anticipated from the Merger,
and we cannot assure you that the Merger will be accretive to us in the near term or at all. Failure to realize such benefits from the
Merger, will result in our pre-Merger stockholders having experienced substantial dilution of their ownership interests in the Company
without receiving any commensurate benefit, or only receiving part of the commensurate benefit to the extent we are able to realize only
part of the strategic and financial benefits currently anticipated from the Merger.
We
may be required to take write-downs or write-offs, restructuring and impairment or other charges that could have a significant negative
effect on our financial condition, results of operations and stock price.
Although
we have conducted due diligence on ENvue Holdings, there can be no assurances that our diligence revealed all material issues that may
be present in ENvue Holdings’ business, that all material issues through a customary amount of due diligence will be uncovered,
or that factors outside of our control will not later arise. As a result, we may be forced to later write-down or write-off assets, restructure
operations, or incur impairment or other charges that could result in losses. Even if due diligence successfully identifies certain risks,
unexpected risks may arise, and previously known risks may materialize in a manner not consistent with our preliminary risk analysis.
Even though these charges may be non-cash items and may not have an immediate impact on liquidity, the fact that we report charges of
this nature could contribute to negative market perceptions about us or our securities. In addition, charges of this nature may make
future financing difficult to obtain on favorable terms or at all.
Global
economic and political instability and conflicts, such as the conflicts in Venezuela, between Russia and Ukraine or in the Middle East,
could adversely affect our business, financial condition or results of operations.
Our
business could be adversely affected by unstable economic and political conditions within the United States and foreign jurisdictions
and geopolitical conflicts, such as the conflicts in Venezuela, between Russia and Ukraine or in the Middle East. While we do not have
any customer or direct supplier relationships in impacted areas at this time, the current military conflict, and related sanctions, as
well as export controls or actions that may be initiated by nations including the United States, the European Union or Russia (e.g.,
potential cyberattacks, disruption of energy flows, etc.) and other potential uncertainties could adversely affect our business and/or
our supply chain, business partners, employees or customers, and interrupt our ability to supply products, or otherwise adversely impact
our business.
Our
operations and financial performance depend on global and regional economic conditions. Inflation, fluctuations in currency exchange
rates, changes in consumer confidence and demand, and weakness in general economic conditions and threats, or actual recessions, could
materially affect our business, results of operations, and financial condition.
Macroeconomic
conditions impact consumer confidence and discretionary spending, which could adversely affect demand for any products we bring to market.
Consumer spending habits are affected by, among other things, inflation, fluctuations in currency exchange rates, weakness in general
economic conditions, threats or actual recessions, pandemics, wars and military actions, levels of employment, wages, debt obligations,
discretionary income, interest rates, volatility in capital, and consumer confidence and perceptions of current and future economic conditions.
Changes and uncertainty can, among other things, drive GPOs, hospitals, nursing homes and other customers towards other options in the
marketplace that may cost less than our products. The recent declines in, or uncertain economic outlooks for, the U.S., European and
certain other international economies has and may continue to adversely affect consumer and healthcare practice spending. The increase
in the cost of fuel and energy, food and other essential items along with elevated interest rates could reduce consumers’ disposable
income, resulting in less discretionary spending for products like ours. Decreases in disposable income and discretionary spending or
change in consumer confidence and spending habits may adversely affect our revenues and operating results.
While
we have not taken on financial obligations from banking institutions and the impact of rising interest rates (in Israel and globally)
on our financing expenses and income has not been significant, inflation continues to adversely impact spending and trade activities
worldwide and we are unable to predict the impacts of higher inflation on global and regional economies. Higher inflation has also increased
domestic and international shipping costs, raw material prices, and labor rates, which could adversely impact the costs of producing,
procuring and shipping any products we bring to market. If similar trends continue our ability to recover these cost increases through
price increases may have limited effectiveness, resulting in downward pressure on our operating results. Attempts to offset cost increases
with price increases could reduce sales, increase customer dissatisfaction or otherwise harm our reputation. Further, we are unable to
predict the impact of efforts by central banks and federal, state and local governments to combat elevated levels of inflation. If their
efforts to reduce inflation are too aggressive, they may lead to a recession. Alternatively, if they are insufficient or are not sustained
long enough to lower inflation to more acceptable levels, consumer spending may be adversely impacted for a prolonged period of time.
Any of these events could materially affect our business and operating results.
If
we are unable to raise additional capital, our clinical trials and product development will be limited and our long-term viability will
be threatened; however, if we do raise additional capital, your percentage ownership as a stockholder could decrease and constraints
could be placed on the operations of our business.
We
have experienced negative operating cash flows since our inception and have funded our operations primarily from proceeds of the sale
of our securities, with only limited revenue being generated from our product sales. In order to fully realize our business objectives,
we may need to raise additional capital. We will seek to raise such additional funds through equity or debt financings, or strategic
alliances with third parties, either alone or in combination with equity financings. These financings could result in substantial dilution
to the holders of our common stock, or require contractual or other restrictions on our operations or on alternatives that may be available
to us. If we raise additional funds by issuing debt securities, these debt securities could impose significant restrictions on our operations
through the imposition of restrictive covenants and requiring us to pledge assets in order to secure repayment. In addition, if we raise
funds through the sale of equity, we may issue equity securities with rights superior to our common stock, including voting rights, rights
to proceeds upon our liquidation or sale, rights to dividends and rights to appoint board members. There can be no assurance that we
will be able to complete a required financing on acceptable terms or at all. If such financing is not available on satisfactory terms,
or is not available in sufficient amounts, we may be required to delay, limit or eliminate the development of business opportunities.
The failure to procure such required financing could have a material adverse effect on our business, financial condition and results
of operations, or threaten our ability to continue as a going concern.
A
variety of factors could impact the timing and amount of any required financings, including, without limitation:
| |
● |
unforeseen
developments during our clinical trials; |
| |
● |
delays
in our receipt of required regulatory approvals; |
| |
● |
delayed
market acceptance of our products; |
| |
● |
unanticipated
expenditures in our acquisition and defense of intellectual property rights, and/or the loss of those rights; |
| |
● |
the
failure to develop strategic alliances for the marketing of some of our product candidates; |
| |
● |
unforeseen
changes in healthcare reimbursement for any of our approved products; |
| |
● |
lack
of financial resources to adequately support our operations; |
| |
● |
difficulties
in maintaining commercial scale manufacturing capacity and capability; |
| |
● |
unanticipated
difficulties in operating in international markets; |
| |
● |
unanticipated
financial resources needed to respond to technological changes and increased competition; |
| |
● |
unforeseen
problems in attracting and retaining qualified personnel; |
| |
● |
enactment
of new legislation or administrative regulations; |
| |
● |
the
application to our business of new regulatory interpretations; |
| |
● |
claims
that might be brought in excess of our insurance coverage; |
| |
● |
the
failure to comply with regulatory guidelines; |
| |
● |
the
uncertainty in industry demand; and |
| |
● |
the
delisting of our common stock from Nasdaq. |
Any
required financing efforts may divert our management from their day-to-day activities, which may adversely affect its ability to develop
and commercialize our products. Moreover, if we complete additional financing by issuing equity securities, the percentage ownership
of its existing stockholders may be reduced, and accordingly these stockholders may experience substantial dilution. Given our need for
cash and that equity issuances are the most common type of fundraising for similarly situated companies, the risk of dilution is particularly
significant for our stockholders.
In
addition, although we have no present commitments or understandings to do so, we may seek to expand our operations and product lines
through acquisitions or joint ventures. Any acquisition or joint venture would likely increase our capital requirements.
If
we fail to obtain an adequate level of reimbursement for our approved products by third party payers, there may be no commercially viable
markets for our approved products or the markets may be much smaller than expected.
The
availability and levels of reimbursement by governmental and other third-party payers affect the market for our commercial products.
The efficacy, safety, performance and cost-effectiveness of our product and product candidates, and of any competing products, will determine
the availability and level of reimbursement. Reimbursement and healthcare payment systems vary significantly by country, and include
both government sponsored healthcare and private insurance. To obtain reimbursement or pricing approval in some countries, we may be
required to produce clinical data, which may involve one or more clinical trials, that compares the cost-effectiveness of our approved
products to other available therapies. We may not obtain reimbursement or pricing approvals in markets we seek to enter in a timely manner,
if at all. Our failure to receive reimbursement or pricing approvals in target markets would negatively impact market acceptance of our
products in these jurisdictions, placing us at a material cost disadvantage to our competitors.
Even
if we obtain reimbursement approvals for our products, we believe that, in the future, reimbursement for any of our products or product
candidates may be subject to increased restrictions both in the United States and in international markets. Future legislation, regulation
or policies of third-party payers that limit reimbursement may adversely affect the demand for our products currently under development
and our ability to sell our products on a profitable basis. In addition, third party payers continually attempt to contain or reduce
the costs of healthcare by challenging the prices charged for healthcare products and services.
In
the United States, specifically, health care providers, such as hospitals and clinics, and individual patients, generally rely on third-party
payers. Third-party reimbursement is dependent upon decisions by the Centers for Medicare and Medicaid Services, contracted Medicare
carriers or intermediaries, individual managed care organizations, private insurers, other governmental health programs and other payers
of health care costs. Failure to receive or maintain favorable coding, coverage and reimbursement determinations for our products by
these organizations could discourage medical practitioners from using or prescribing our products due to their costs. In addition, with
recent federal and state government initiatives directed at lowering the total cost of health care, the U.S. Congress and state legislatures
will likely continue to focus on health care reform including the reform of the Medicare and Medicaid programs, and on the cost of medical
products and services, which could limit reimbursement. Additionally, third-party payers are increasingly challenging the prices charged
for medical products and services, and imposing conditions on payment. We may be unable to sell our products on a profitable basis if
third-party payers deny coverage, provide low reimbursement rates or reduce their current levels of reimbursement.
The
medical device and therapeutic product industries are highly competitive and subject to rapid technological change. If our competitors
are able to develop and market products that are safer and more effective than any products we may develop, our commercial opportunities
will be reduced or eliminated.
Our
success depends, in part, upon our ability to maintain a competitive position in the development of technologies and products. We face
competition from established medical device companies, such as (i) with respect to our NanoVibronix business, Neurometrix Inc., Zetrox
(a subsidiary of the 3M Company), Smith & Nephew plc, manufacturers of certain portable ultrasound devices capable of self-administered
use and (ii) with respect to ENvue Holdings business, Cardinal Health and Avanos Medical, as well as from academic institutions, government
agencies, and private and public research institutions in the United States and abroad. Most, if not all, of our principal competitors
have significantly greater financial resources and expertise than we do in research and development, manufacturing, pre-clinical testing,
conducting clinical trials, obtaining regulatory approvals, marketing approved products, protecting and defending their intellectual
property rights and designing around the intellectual property rights of others. Other small or early-stage companies may also prove
to be significant competitors, particularly through collaborative arrangements, or mergers with, or acquisitions by, large and established
companies, or through the development of novel products and technologies.
The
industry in which we operate has undergone, and we expect it to continue to undergo, rapid and significant technological change, and
we expect competition to intensify as technological advances are made. Our competitors may be able to respond to changes in technology
or the marketplace faster than us. Our competitors may develop and commercialize medical devices that are safer or more effective or
are less expensive than any products that we may develop. We also compete with our competitors in recruiting and retaining qualified
scientific and management personnel, in establishing clinical trial sites and patient registration for clinical trials, and in acquiring
technologies complementary to our programs or advantageous to our business. Given our small size and lack of resources, we are often
at a disadvantage with our competitors in all of these areas, which could limit or eliminate our commercial opportunities.
We
face the risk of product liability claims and may not be able to obtain insurance.
Our
business exposes us to the risk of product liability claims that are inherent in the development of medical devices and products. If
the use of one or more of our products harms people, we may be subject to costly and damaging product liability claims brought against
us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products. We currently
carry clinical trial and product liability insurance for the products we sell. However, we cannot predict all of the possible harms or
side effects that may result and, therefore, the amount of insurance coverage we hold may not be adequate to cover all liabilities we
might incur. We intend to expand our insurance coverage to include the sale of additional commercial products as we obtain marketing
approval for our product candidates in development and as our sales expand, but we may be unable to obtain commercially reasonable product
liability insurance for such products. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential
product liability claims and we continue to make sales, or if our coverages turns out to be insufficient, we may be exposed to significant
liabilities, which may materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused
by our products and do not have sufficient insurance coverage, our liability could exceed our total assets and our ability to pay the
liability. A product liability claim or series of claims brought against us would decrease our cash and could reduce our value or marketability.
Our
products may not be developed or commercialized successfully.
Our
products are based on technologies that have not been used previously in the manner we propose and must compete with more established
treatments currently accepted as the standards of care. Market acceptance of our products will largely depend on our ability to demonstrate
their relative safety, efficacy, cost-effectiveness and ease of use.
We
are subject to the risks that:
| ● |
the
FDA or a foreign regulatory authority finds our products ineffective or unsafe; |
| ● |
we
do not receive necessary regulatory approvals; |
| ● |
the
regulatory review and approval process may take much longer than anticipated, requiring additional time, effort and expense to respond
to regulatory comments and/or directives; |
| ● |
we
are unable to get our products in commercial quantities at reasonable costs; and |
| ● |
the
patient and physician community does not accept our products. |
In
addition, our product development program may be curtailed, redirected, eliminated or delayed at any time for many reasons, including:
| ● |
adverse
or ambiguous results; |
| ● |
undesirable
side effects that delay or extend the trials; |
| ● |
the
inability to locate, recruit, qualify and retain a sufficient number of clinical investigators or patients for our trials; and |
| ● |
regulatory
delays or other regulatory actions. |
Additionally,
we currently have limited experience in marketing or selling our products, and we have a limited marketing and sales staff and distribution
capabilities. Developing a marketing and sales force is time-consuming and will involve the investment of significant amounts of financial
and management resources, and could delay the launch of new products or expansion of existing product sales. In addition, we compete
with many companies that currently have extensive and well-funded marketing and sales operations. If we fail to establish successful
marketing and sales capabilities or fail to enter into successful marketing arrangements with third parties, our ability to generate
revenues will suffer.
Furthermore,
even if we enter into marketing and distributing arrangements with third parties, we may have limited or no control over the sales, marketing
and distribution activities of these third parties, and these third parties may not be successful or effective in selling and marketing
our products. If we fail to create successful and effective marketing and distribution channels, our ability to generate revenue and
achieve our anticipated growth could be adversely affected. If these distributors experience financial or other difficulties, sales of
our products could be reduced, and our business, financial condition and results of operations could be harmed.
We
cannot predict whether we will successfully develop and commercialize our products. If we fail to do so, we will not be able to generate
substantial revenues, if any.
We
are highly dependent on our senior management team and key personnel, and our business could be harmed if we are unable to attract and
retain personnel necessary for our success.
We
are highly dependent on our senior management and key personnel. Our success will depend on our ability to retain senior management and
to attract and retain qualified personnel in the future, including sales and marketing professionals, engineers, scientists, data science
specialists and other highly skilled personnel and to integrate current and additional personnel in all departments.
Competition
for skilled personnel in our market is intense and may limit our ability to hire and retain highly qualified personnel on acceptable
terms, or at all. We face intense competition in our hiring efforts from other medical device companies, as well as from universities
and nonprofit research organizations, and we may have to pay higher salaries to attract and retain qualified personnel. We are also at
a disadvantage in recruiting and retaining key personnel as our small size and limited resources may be viewed as providing a less stable
environment, with fewer opportunities than would be the case at one of our larger competitors. The loss of one or more of our senior
management or key personnel, or our inability to attract additional qualified personnel, could substantially impair our ability to implement
our business plan. In addition, the replacement of key personnel likely would involve significant time and costs, and may significantly
delay or prevent the achievement of our business objectives.
To
induce valuable employees to remain at our company, in addition to salary and cash incentives, we have issued stock options that vest
over time, restricted share units subject to vesting conditions, and certain performance warrants. The value to employees of stock options
that vest over time may be significantly affected by fluctuations in our stock price that are beyond our control, and may at any time
be insufficient to counteract more lucrative offers from other companies. Despite our efforts to retain valuable employees, members of
our management and other key personnel may terminate their employment with us on short notice. Our employment arrangements with our employees
provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice.
We also do not maintain “key man” insurance policies on the lives of these individuals or the lives of any of our other employees.
As
we engage with GPOs and target medical providers to increase adoption of and utilization of the ENvue System, expand our product offerings
in the future and increase our future marketing efforts, we will need to build and expand the reach of our marketing and sales networks.
Our future success will depend largely on our ability to continue to hire, train, retain and motivate skilled employees with significant
technical knowledge in various areas. An inability to attract, hire, train and retain employees will harm our sales, business, financial
condition, and results of operations.
Our
need to increase the size of our organization in order to successfully manage our growth may adversely affect our business, financial
condition and results of operations.
We
have a small number of planned employees, and our management systems currently in place are not likely to be adequate to support our
future growth plans. Our ability to grow and to manage our growth effectively will require us to hire, train, retain, manage and motivate
additional employees and to implement and improve our operational, financial and management systems. These demands also may require the
hiring of additional senior management personnel or the development of additional expertise by our senior management personnel. Hiring
a significant number of additional employees, particularly those at the management level, would increase our expenses significantly.
Moreover, if we fail to expand and enhance our operational, financial and management systems in conjunction with our potential future
growth, such failure could have a material adverse effect on our business, financial condition and results of operations.
We
face risks associated with litigation and claims.
We
may, in the future, be involved in one or more lawsuits, claims or other proceedings. These suits could concern issues including contract
disputes, employment actions, employee benefits, taxes, environmental, health and safety, fraud and abuse, personal injury and product
liability matters.
On
February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the
International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach
of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges,
in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of
an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million. The Company vigorously defended the claims
asserted by Protrade.
On
March 15, 2022, the arbitrator issued a final award, which, determined that (i) the Company had the right to terminate the Exclusive
Distribution Agreement; (ii) the Company did not breach the duty of good faith and fair dealing with regard to the Exclusive Distribution
Agreement; and (iii) the Company did not breach any confidentiality obligations to Protrade. Nevertheless, the arbitrator determined
that the Company did not comply with the obligation to supply Protrade with a year’s supply of patches, and awarded Protrade $1,500,250,
which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the
Company allegedly failed to supply Protrade with certain patches utilized by users of DV0057 Painshield MD device. The arbitrator based
the decision on the testimony of Protrade’s president who asserted that a user would use in excess of 33 patches per each device.
The Company believes that the number of patches per device alleged by Protrade is grossly inflated, and that these claims were not properly
raised before the arbitrator. Accordingly, on April 13, 2022, the Company submitted an application for the correction of the award which
the arbitrator denied on June 22, 2022.
On
April 5, 2022, Protrade filed a Petition with the Supreme Court of New York Nassau County seeking to confirm the Award. On April 13,
2022, the Company submitted an application to the ICA seeking to correct an error in the award based on the evidence that the Company
only sold 2-3 reusable patches per device contrary to the 33 reusable patches claimed by Protrade. The same arbitrator who issued the
award, denied the application.
On
July 22, 2022, the Company filed a cross-motion seeking to vacate arbitration award on the grounds that the arbitrator exceeded her authority,
that the award was procured by fraud, and that the arbitrator failed to follow procedures established by New York law. In particular,
the Company averred in its motion that Protrade’s witness made false statements in arbitration, and that the arbitrator resolved
a claim that was never raised by Protrade and that has no factual basis.
On
October 3, 2022, the court issued a decision granting Protrade its petition to confirm the award and denying the cross-motion.
On
November 9, 2022, the Company filed a motion to re-argue and renew its cross-motion to vacate the arbitration decision based on newer
information that was not available during the initial hearing. On the same day, the Company also filed a notice of appeal with the Appellate
Division, Second Department. On March 21, 2023, the court denied the motion to re-argue and renew.
On
July 10, 2023, the Company filed its appeal with the Appellate Division, Second Department. That appeal is now fully briefed. In February
2025, the Second Department informed counsel for the Company that the Second Department was beginning to process the appeal for calendaring.
As
of December 31, 2025, and 2024, the Company accrued the amount of the arbitration award to Protrade of approximately $2.3 and $2.1 million,
respectively, including interest which is classified in “Other accounts payable and accrued expenses”.
On
March 30, 2026, the Appellate Division of the Second Department issued a decision and order which affirmed the judgment of the
Supreme Court, Nassau County in its entirety and dismissed the appeal, stating inter alia that “NanoVibronix
failed to establish that the arbitration award violated a strong public policy, was irrational, procured by fraud, or clearly
exceeded a specifically enumerated limitation of the arbitrator’s power”. It further held that NanoVibronix
had not demonstrated that it had shown sufficient new facts to the court on the motion to renew. The decision was conclusory and did
not analyze the facts as argued by NanoVibronix nor did it distinguish them
specifically. The Company is currently reviewing the decision and considering its
available alternatives.
Our
products and information technology systems are critical to our business. Issues with product development or enhancements, IT system
integration, implementation, updates and upgrades could disrupt our operations and have a material impact on our business and operating
results.
We
rely on the efficient, uninterrupted and secure operation of our IT systems and are dependent on key third-party software embedded in
our products and IT systems as well as third-party hosted IT systems to support our operations. All software and IT systems are vulnerable
to damage, cyber-attacks or interruption from a variety of sources. To effectively manage and improve our operations, our IT systems
and applications require an ongoing commitment of significant expenditures and resources to maintain, protect, upgrade, enhance and restore
existing systems and develop new systems to keep pace with continuing changes in information processing technology, evolving industry
and regulatory standards, increasingly sophisticated cyber threats, and changing consumer preferences. Failure to adequately protect
and maintain the integrity of our products and IT systems may result in a material effect on our financial position, results of operations
and cash flows.
We
plan to continuously upgrade and issue new releases of our products and customer-facing software applications, upon which our operations
depend. Software applications and products containing software frequently contain errors or defects, especially when first introduced
or when new versions are released. Additionally, the third-party software integrated into or interoperable with our products and services
will routinely reach end of life, and as a consequence, may be exposed to additional vulnerabilities, including increased security risks,
errors and malfunctions that may be irreparable or difficult to repair. The discovery of a defect, error or security vulnerability in
our products, software applications or IT systems, incompatibility with future customers’ computer operating systems and hardware
configurations with a new release or upgraded version or the failure of our products or primary IT systems may cause adverse consequences,
including: delay or loss of revenues, significant remediation costs, delay in market acceptance, loss of data, disclosure of financial,
health or other personal information of any customers or patients, product recalls, damage to our reputation, or increased service costs,
any of which could have a material effect on our business, financial condition or results of our operations and the operations of our
potential customers or our business partners.
Our
business and operations would suffer in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security.
In
the ordinary course of our business, we collect and store sensitive data, including intellectual property, research data, our proprietary
business information and that of our suppliers, technical information about our products, clinical trial plans and employee records.
Similarly, our third-party providers possess certain of our sensitive data and confidential information. The secure maintenance of this
information is critical to our operations and business strategy. Despite the implementation of security measures, our internal computer
systems, and those of third parties on which we rely, are vulnerable to damage from computer viruses, malware, ransomware, cyber fraud,
natural disasters, terrorism, war, telecommunication and electrical failures, cyber-attacks or cyber-intrusions over the Internet, attachments
to emails, persons inside our organization, or persons with access to systems inside our organization. The risk of a security breach
or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists,
has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased.
Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, encrypted, lost
or stolen. Any such access, inappropriate disclosure of confidential or proprietary information or other loss of information, including
our data being breached at third-party providers, could result in legal claims or proceedings, liability or financial loss under laws
that protect the privacy of personal information, disruption of our operations or our product development programs and damage to our
reputation, which could adversely affect our business.
We
may acquire businesses or products, or form strategic alliances, in the future, and may not realize the benefits of such acquisitions.
We
may acquire additional businesses or products, form strategic alliances, or create joint ventures with third parties that we believe
will complement or augment our existing business. If we acquire businesses with promising markets or technologies, we may not be able
to realize the benefit of acquiring such businesses if we are unable to successfully integrate them with our existing operations and
company culture. We may encounter numerous difficulties in developing, manufacturing, and marketing any new products resulting from a
strategic alliance or acquisition that delay or prevent us from realizing their expected benefits or enhancing our business. There is
no assurance that, following any such acquisition, the combined company will achieve the synergies expected to justify the transaction,
which could result in a material adverse effect on our business and prospects.
Certain
stockholders could attempt to influence changes within the Company, which could adversely affect our operations, financial condition
and the value of our common stock.
Our
stockholders may from time to time seek to acquire a controlling stake in the Company, engage in proxy solicitations, advance stockholder
proposals or otherwise attempt to effect changes. Campaigns by stockholders to effect changes at publicly traded companies are sometimes
led by investors seeking to increase short-term stockholder value through actions such as financial restructuring, increased debt, special
dividends, stock repurchases or sales of assets or the entire company. Responding to proxy contests and other actions by activist stockholders
can be costly and time-consuming. These actions could adversely affect our operations, financial condition, and the value of our common
stock.
Our
operations may be impacted by natural disasters, which may become more frequent or severe as a result of climate change and may adversely
impact our business and operating results as well as those of our potential customers and suppliers.
Natural
disasters can impact us and our potential customers, as well as suppliers critical to our operations. Natural disasters include earthquakes,
tsunamis, floods, droughts, hurricanes, wildfires, and other extreme weather conditions that can cause deaths, injuries, and critical
health crises, power outages, restrictions and shortages of food, water, shelter, and medical supplies, telecommunications failures,
materials scarcity, price volatility and other ramifications. Climate change is likely to increase both the frequency and severity of
natural disasters and, consequently, risks to our business and operations.
The
effects of climate change on regional and global economies could change the supply, demand or availability of sources of energy or other
resources material to our products and operations and affect the availability or cost of natural resources and goods and services on
which we and our suppliers rely.
Risks
Related to Our Regulatory and Compliance Matters
Complying
with regulations enforced by FDA and other regulatory authorities is expensive and time consuming, and failure to comply could result
in substantial penalties.
We
are subject to extensive governmental regulation, including the requirement of U.S. Food and Drug Administration approval or clearance
before NanoVibronix product candidates may be marketed and after approval or clearance and during the marketing of our products. The
process of obtaining FDA approval is lengthy, expensive and uncertain, and we cannot be sure that our additional product candidates will
be approved in a timely fashion, or at all. If the FDA does not approve or clear our product candidates in a timely fashion, or at all,
our business and financial condition would likely be adversely affected.
Both
before and after approval or clearance of our product candidates, we, our product candidates, our suppliers and our contract manufacturers
are subject to extensive regulation by governmental authorities in the United States and other countries. Failure to comply with applicable
requirements could result in, among other things, any of the following actions:
| ● |
FDA
issuance of Form 483 or Warning Letters, which may be made public and may lead to further regulatory or enforcement actions, or similar
letters by other regulatory authorities; |
| ● |
fines
and other monetary penalties; |
| ● |
unanticipated
expenditures; |
| ● |
delays
in FDA approval and clearance, or FDA refusal to approve or clear a product candidate; |
| ● |
product
recall or seizure; |
| ● |
interruption
of manufacturing or clinical trials; |
| ● |
operating
restrictions; |
| ● |
injunction
or other restrictions imposed on our operations, including closing our facilities or our contract manufacturers’ facilities;
or |
| ● |
criminal
prosecutions. |
In
addition to the approval and clearance requirements, numerous other regulatory requirements apply, both before and after approval or
clearance, to us, our products and product candidates, and our suppliers and contract manufacturers. These include requirements related
to the following:
| ● |
testing
and quality control; |
| ● |
manufacturing; |
| ● |
quality
assurance; |
| ● |
labelling; |
| ● |
advertising; |
| ● |
promotion
(including the prohibition on promoting devices for “off-label” uses); |
| ● |
distribution; |
| ● |
export; |
| ● |
reporting
to the FDA certain adverse experiences associated with the use of the products, as well as our discovery of defects or a product’s
failure to comply with design specifications or applicable law; and |
| ● |
obtaining
additional approvals or clearances for certain modifications to the products or their labelling or claims. |
| |
|
We
are also subject to inspection by the FDA to determine our compliance with regulatory requirements, as are our suppliers and contract
manufacturers, and we cannot be sure that the FDA will not identify compliance issues that may disrupt production or distribution, or
require substantial resources to correct. We also cannot be sure that the FDA will agree with our analysis of, conclusions regarding,
or handling of various situations that arise with our products. If it is determined that we failed to comply with any of our regulatory
obligations, we could be subject to a wide range of enforcement actions that could limit our ability to continue to successfully commercialize
impacted products or otherwise adversely impact us.
The
FDA’s requirements may change and additional government regulations may be promulgated that could affect us, our product candidates,
and our suppliers and contract manufacturers. We cannot predict the likelihood, nature or extent of government regulation that may arise
from future legislation or administrative action. There can be no assurance that we will not be required to incur significant costs to
comply with such laws and regulations in the future, or that such laws or regulations will not have a material adverse effect upon our
business.
In
addition, ENvue Holdings’ product, the ENvue System (for which we have obtained FDA 510(k) clearance) is, and future ENvue Holdings
products can be considered medical devices and, accordingly, are subject to rigorous regulation by government agencies in the U.S. and
other countries in which we intend to sell our products. Compliance with these rigorous regulations will affect capital expenditures,
earnings and the competitive position of the Company. These regulations vary from country to country but cover, among other things, the
following activities with respect to medical devices:
| ● |
design,
development and manufacturing; |
| |
|
| ● |
testing,
labeling, content and language of instructions for use and storage; |
| |
|
| ● |
product
storage and safety; |
| |
|
| ● |
marketing,
sales and distribution; |
| |
|
| ● |
pre-market
clearance or approval; |
| |
|
| ● |
record
keeping procedures; |
| |
|
| ● |
advertising
and promotion; |
| |
|
| ● |
recalls
and field safety corrective actions; |
| ● |
post-market
surveillance; |
| |
|
| ● |
post-market
approval studies; and |
| |
|
| ● |
product
import and export. |
The
regulations to which we are subject are complex. Regulatory changes could result in restrictions on our ability to carry on or expand
our operations, higher than anticipated costs, or lower than anticipated sales. Our failure to comply with applicable regulatory requirements
could result in enforcement action by FDA or state agencies, which may include any of the following sanctions:
| ● |
warning
letters, fines, injunctions, consent decrees, and civil penalties; |
| |
|
| ● |
repair,
replacement, refunds, recall, or seizure of our products; |
| |
|
| ● |
operating
restrictions or partial suspension or total shutdown of production; |
| |
|
| ● |
refusing
our requests for 510(k) clearance or pre-market approval of new products, new intended uses, or modifications to existing products; |
| |
|
| ● |
withdrawing
clearance or pre-market approvals that have already been granted; and |
| |
|
| ● |
criminal
prosecution. |
If
any of these events were to occur, they could harm our business.
UroShield
has not been cleared or approved by the FDA, nor has it undergone the same type of review as an FDA-approved or cleared device.
Before
we can sell a new medical device in the U.S., or market a new use of, new claim for, or significant modification to a legally marketed
device, we must first obtain either FDA 510(k) clearance or pre-market approval, unless an exemption applies. In the 510(k) clearance
process, before a device may be marketed, the applicant must submit a premarket notification to FDA under Section 510(k) of the FD&C
Act, and FDA must determine that a proposed device is “substantially equivalent” to a legally-marketed “predicate”
device. To be “substantially equivalent,” the proposed device must have the same intended use as the predicate device, and
either have the same technological characteristics as the predicate device or have different technological characteristics, not raise
different questions of safety or effectiveness than the predicate device, and be as safe and as effective as the predicate device. The
510(k) clearance process can be expensive and uncertain and typically takes from three to 12 months, but may last significantly longer.
Clinical data may be required in connection with an application for 510(k) clearance. Furthermore, even if we are granted regulatory
clearances, they may include limitations on the indications for use or intended uses of the device, which may limit the market for the
device.
Our
ENvue System is a Class II medical device and received FDA clearance under the 510(k) pathway for marketing to adults (ages 22 and above)
only. FDA can delay, limit, or deny 510(k) clearance, or approval or reclassification, of a device for many reasons, including:
| ● |
we
may be unable to demonstrate to FDA’s satisfaction that the product candidate or modifications are substantially equivalent
to a proposed predicate device or safe and effective for their intended uses; |
| |
|
| ● |
we
may be unable to demonstrate that the clinical and other benefits of the device outweigh the risks; and |
| |
|
| ● |
the
applicable regulatory authority may identify deficiencies in our submissions or in the facilities
or processes of our third party contract manufacturers.
|
Any
delay or failure to obtain necessary regulatory clearances or approvals could harm our business. For example, if we decide to market
the ENvue System for a broader or additional indication(s) for use and/or make any material modifications to any element of the device
and/or the manufacturing or distribution thereof in the future, an additional 510(k) submission, and FDA clearance thereof, will be required
prior to making any promotional communications expressly or impliedly claiming that the device may be used for such indication(s) and/or
prior to making such modification, respectively.
In
addition, FDA may change its policies, adopt additional regulations, revise existing regulations, or take other actions, or Congress
may enact different or additional statutory requirements, which may prevent or delay clearance of our future products under development
or impact our ability to modify our currently marketed products on a timely basis. Such policy, statutory, or regulatory changes could
impose additional requirements upon us that could delay our ability to obtain new 510(k) clearances, increase the costs of compliance,
or restrict our ability to maintain our current marketing authorizations.
We
received our European CE mark, indicating that we affirm our product’s conformity with European health, safety and environmental
protection standards, in 2021. We will also need to obtain regulatory approval in other foreign jurisdictions in which we plan to market
and sell our products. The time required to obtain registrations or approvals, if required by other countries, may be longer than that
required for FDA clearance, and requirements for such registrations, clearances, or approvals may significantly differ from FDA requirements.
If we modify our products, we may need to apply for additional regulatory approvals before we are permitted to sell the modified product.
In addition, we may not continue to meet the quality and safety standards required to maintain the authorizations that we have received.
If we are unable to maintain our authorizations in a particular country, we will no longer be able to sell the applicable product in
that country.
Failure
to comply with these rules, regulations, self-regulatory codes, circulars, and orders could result in significant civil and criminal
penalties and costs and could have a material adverse impact on our business. Also, these regulations may be interpreted or applied by
a prosecutorial, regulatory, or judicial authority in a manner that could require us to make changes in our operations or incur substantial
defense and settlement expenses. Even unsuccessful challenges by regulatory authorities or private relators could result in reputational
harm and the incurring of substantial costs. In addition, many of these laws are vague or indefinite and have not been interpreted by
the courts and have been subject to frequent modification and varied interpretation by prosecutorial and regulatory authorities, increasing
compliance risks.
Certain
modifications to our products may require new 510(k) clearance or other marketing authorizations.
Once
a medical device is permitted to be legally marketed in the U.S. pursuant to a 510(k) clearance, a medical device developer may be required
to notify FDA of certain modifications to the device. Medical device developers determine in the first instance whether a change to a
product requires a new premarket submission, but FDA may review any such decision.
While
our ENvue System received 510(k) clearance in February 2019, we may in the future apply for 510(k) clearance for updated components of
our ENvue System, which must, then, be found by the FDA to be substantially equivalent to the cleared ENvue System and, thus, may not
be lawfully marketed in the U.S. until the FDA makes a substantial equivalence determination and issues the requisite 510(k) clearance
for the updated ENvue System. Although the development of our ENvue System has been carefully monitored and documented by professionals
who are experienced in the FDA clearance process, there is no assurance that the FDA will agree that an updated component of our ENvue
System is substantially equivalent to the cleared ENvue System and allow the updated ENvue System to be marketed in the United States.
The FDA may determine that the device is not substantially equivalent and require a premarket approval (“PMA”) or, more likely,
a de novo reclassification, and/or require further information, such as additional test data, including data from additional clinical
studies, before it is able to make a determination regarding substantial equivalence or PMA. By requesting additional information, the
FDA can delay market introduction of an updated ENvue System and increase the resources needed to gain clearance or PMA. Delays in receipt
of or failure to receive any necessary 510(k) clearance, de novo classification, or PMA, or the imposition of stringent restrictions
for our ENvue System, could have a material adverse effect on our business, results of operations and financial condition.
In
the future, we may make other modifications to our products, including our ENvue System, and determine, based on our review of the applicable
FDA regulations and guidance, that in certain instances new 510(k) clearances or other premarket submissions are not required. If FDA
disagrees with our determinations, we may be subject to a wide range of enforcement actions, including, for example, a warning letter,
among other consequences, after which we will likely have to cease marketing the applicable modified product and/or to recall distributed
units of such modified product until we obtain the requisite clearance or approval.
Failure
to obtain regulatory approval in foreign jurisdictions will prevent us from marketing our products abroad.
International
sales of our products and any of our products that we commercialize are subject to the regulatory requirements of each country in which
the products are sold. Accordingly, the introduction of our products in markets outside the United States where we do not already possess
regulatory approval will be subject to regulatory approvals in those jurisdictions. The regulatory review process varies from country
to country. Many countries impose product standards, packaging and labelling requirements, and import restrictions on medical devices.
In addition, each country has its own tariff regulations, duties and tax requirements, as well as reimbursement and healthcare payment
systems. The approval by foreign government authorities is unpredictable and uncertain, and can be expensive. We may be required to perform
additional pre-clinical, clinical or post-approval studies even if FDA approval has been obtained. Our ability to market our approved
products could be substantially limited due to delays in receipt of, or failure to receive, the necessary approvals or clearances.
To
the extent we intend to sell medical devices in member states of the European Union (“EU”), our products must comply with
the general safety and performance requirements of the Medical Devices Regulation (“MDR”) (Regulation (EU) No 2017/745),
which repeals and replaces the Medical Devices Directive (the “MDD”). Compliance with these requirements is a prerequisite
to be able to affix the European conformity (“CE” or “CE mark”) to our products, without which they cannot be
sold or marketed in the EU. All medical devices placed on the market in the EU must meet the general safety and performance requirements
laid down in Annex I to the MDR, including the requirement that a medical device must be designed and manufactured in such a way that,
during normal conditions of use, it is suitable for its intended purpose. Medical devices must be safe and effective and must not compromise
the clinical condition or safety of patients, or the safety and health of users and - where applicable - other persons, provided that
any risks which may be associated with their use constitute acceptable risks when weighed against the benefits to the patient and are
compatible with a high level of protection of health and safety, taking into account the generally acknowledged state of the art. To
demonstrate compliance with the general safety and performance requirements, we must undergo a conformity assessment procedure, which
varies according to the type of medical device and its (risk) classification. Except for low risk medical devices (Class I), where the
manufacturer can self-assess the conformity of its products with the general safety and performance requirements (except for any parts
which relate to sterility, metrology or reuse aspects), a conformity assessment procedure requires the intervention of a notified body.
Notified bodies are independent organizations designated by EU member states to assess the conformity of certain products before being
placed on the market.
We
are uncertain regarding the success of our clinical trials for our products in development.
There
can be no assurance that we will be able to successfully complete the U.S. and foreign regulatory approval processes for
products in development. In addition, there can be no assurance that we will not encounter additional problems that will cause us to
delay, suspend or terminate our clinical trials. In addition, we cannot make any assurance that clinical trials will be deemed sufficient
in size and scope to satisfy regulatory approval requirements, or, if completed, will ultimately demonstrate our products to be safe
and efficacious.
Healthcare
reform measures could adversely affect our business and financial results.
In
the United States, there have been, and we expect there will continue to be, a number of legislative and regulatory changes to the healthcare
system in ways that may adversely affect our business and financial results. Federal and state lawmakers regularly propose and, at times,
enact legislation that could result in significant changes to the healthcare system, some of which are intended to contain or reduce
the costs of medical products and services. Current and future legislative proposals to further reform healthcare or reduce healthcare
costs may limit coverage of or lower reimbursement for our products. The cost containment measures that payers and providers are instituting
and the effect of any healthcare reform initiative implemented in the future could impact our revenue from the sale of our products.
For example, the Patient Protection and Affordable Act of 2010, commonly referred to as the Affordable Care Act, contains a number of
provisions, including those governing enrollment in federal healthcare programs, reimbursement changes and fraud and abuse measures,
all of which will impact existing government healthcare programs and will result in the development of new programs.
There
have been executive, judicial and Congressional challenges to certain aspects of the Affordable Care Act for over a decade. However,
as of the Supreme Court’s ruling ordering the dismissal of, arguably, the most promising case challenging the Affordable Care Act
to-date in June 2021, it appears that the Affordable Care Act will remain in-effect in its current form for the foreseeable future. We
cannot predict what additional challenges to the Affordable Care Act may arise in the future, the outcome thereof, or the impact any
such actions may have on our business. Additionally, the Biden administration has introduced various measures in recent years, focusing
on healthcare and medical-product pricing, in particular. It remains to be seen how these measures will affect our business and there
is uncertainty as to what other healthcare programs and regulations may be implemented or changed at the federal and/or state level in
the U.S., but it is possible that such initiatives could have an adverse effect on our ability to obtain FDA approval or clearance and/or
successfully commercialize products in the U.S. in the future. For example, any changes that reduce, or impede the ability of healthcare
providers to obtain reimbursement for medical procedures in which the products we currently, or intend to, commercialize are used, or
that reduce medical procedure volumes, could adversely affect our operations and/or future business plans. The financial impact of U.S.
healthcare reform legislation over the next few years will depend on a number of factors, including the policies reflected in implementing
regulations and guidance and changes in sales volumes for medical devices affected by the legislation. From time to time, legislation
is drafted, introduced, and passed that could significantly change the statutory provisions governing coverage, reimbursement, pricing,
and marketing of medical device products. In addition, third-party payor coverage and reimbursement policies are often revised or interpreted
in ways that may significantly affect our business and our products.
On
April 5, 2017, the European Parliament passed the MDR which repeals and replaces the EU Medical Device Directive and became effective
on May 26, 2021. The Medical Devices Regulation, among other things, is intended to establish a uniform, transparent, predictable, and
sustainable regulatory framework across the EEA for medical devices and ensure a high level of safety and health while supporting innovation.
The new regulations, among other things:
| ● |
strengthen
the rules on placing devices on the market and reinforce surveillance once they are available; |
| |
|
| ● |
establish
explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices
placed on the market; |
| ● |
improve
the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number; |
| |
|
| ● |
set
up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available
in the European Union; and |
| |
|
| ● |
strengthen
rules for the assessment of certain high-risk devices, such as implants, which may have to undergo an additional check by experts
before they are placed on the market. |
These
modifications may have an effect on the way we conduct our business in the EEA.
Any
change in the laws or regulations that govern the clearance and approval processes relating to our current, planned and future products
could make it more difficult and costly to obtain clearance or approval for new products or to produce, market and distribute existing
products. Significant delays in receiving clearance or approval or the failure to receive clearance or approval for our new products
would have an adverse effect on our ability to expand our business.
We
may be subject to certain federal, state, and foreign fraud and abuse laws, health information privacy and security laws, and transparency
laws, which, if violated, could subject us to substantial penalties. Additionally, any challenge to or investigation into our practices
under these laws could cause adverse publicity and be costly to respond to, and thus could harm our business.
There
are numerous U.S. federal and state, as well as foreign, laws pertaining to healthcare fraud and abuse, including anti-kickback, false
claims, and physician transparency laws. Efforts to ensure that our business arrangements with third parties will comply with applicable
healthcare laws and regulations may involve substantial costs. Our business practices and relationships with providers and patients are
subject to scrutiny under these laws. We may also be subject to patient information privacy and security regulation by both the federal
government and the states and foreign jurisdictions in which we conduct our business. The healthcare laws and regulations that may affect
our ability to operate include:
| ● |
the
federal healthcare Medicare and Medicaid Patient Protection Act of 1987 (the “Anti-Kickback Statute”), which prohibits,
among other things, persons, and entities from knowingly and willfully soliciting, offering, receiving or providing remuneration,
directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, lease,
order, or arrange for or recommend a good or service, for which payment may be made, in whole or in part, under federal healthcare
programs, such as Medicare and Medicaid. The term “remuneration” has been broadly interpreted to include anything of
value. The government can establish a violation of the Anti-Kickback Statute without proving that a person or entity had actual knowledge
of the law or a specific intent to violate. Moreover, the government may assert that a claim including items or services resulting
from a violation of the federal healthcare Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal
civil False Claims Act. Although there are a number of statutory exceptions and regulatory safe harbors to the federal healthcare
Anti-Kickback Statute protecting certain common business arrangements and activities from prosecution or regulatory sanctions, the
exceptions and safe harbors are drawn narrowly. Practices that involve remuneration to those who prescribe, purchase, or recommend
medical device products, including discounts, or engaging individuals as speakers, consultants, or advisors, may be subject to scrutiny
if they do not fit squarely within an exception or safe harbor. Our practices may not in all cases meet all of the criteria for safe
harbor protection from anti- kickback liability. Moreover, there are no safe harbors for many common practices, such as reimbursement
support programs, educational or research grants, or charitable donations; |
| ● |
the
federal civil False Claims Act, which prohibits, among other things, individuals or entities from knowingly presenting, or causing
to be presented, false or fraudulent claims for payment of federal government funds, and knowingly making, using or causing to be
made or used a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay
money to the federal government. Private individuals, commonly known as “whistleblowers,” can bring civil False Claims
Act qui tam actions, on behalf of the government and such individuals and may share in amounts paid by the entity to the government
in recovery or settlement. False Claims Act liability is potentially significant in the healthcare industry because the statute provides
for treble damages and serious mandatory penalties for each false or fraudulent claim or statement. The government may assert that
a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent
claim under the federal civil False Claims Act. Many pharmaceutical and medical device manufacturers have been investigated and have
reached substantial settlements under the federal civil False Claims Act in connection with alleged off-label promotion of their
products and allegedly providing free products to customers with the expectation that the customers would bill federal health care
programs for the product. In addition, manufacturers can be held liable under the federal civil False Claims Act even when they do
not submit claims directly to government payers if they are deemed to “cause” the submission of false or fraudulent claims.
There are also criminal penalties, including imprisonment and criminal fines, for making or presenting false, fictitious or fraudulent
claims to the federal government; |
| ● |
the
Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created additional federal criminal statutes
that prohibit, among other things, knowingly and willfully executing or attempting to execute a scheme to defraud any healthcare
benefit program, including private third-party payers, knowingly and willfully embezzling or stealing from a healthcare benefit program,
willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering
up a material fact or making any materially false, fictitious or fraudulent statements or representations, or making or using any
false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection
with the delivery of, or payment for, healthcare benefits, items or services. Similar to the federal healthcare Anti-Kickback Statute,
a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation; |
| |
|
| ● |
the
federal Physician Payments Sunshine Act under the Affordable Care Act, which requires certain manufacturers of drugs, devices, biologics
and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program (with
certain exceptions) to report annually to the United States Department of Health and Human Services, Centers for Medicare and Medicaid
Services, information related to payments and other transfers of value to physicians (defined to include doctors, dentists, optometrists,
podiatrists and chiropractors) and teaching hospitals, and applicable manufacturers and group purchasing organizations, as well as
ownership and investment interests held by physicians and their immediate family members. Since January 2022, applicable manufacturers
are also required to report information regarding payments and transfers of value provided to physician assistants, nurse practitioners,
clinical nurse specialists, certified nurse anesthetists, and certified nurse-midwives; |
| |
|
| ● |
HIPAA,
as amended by Health Information Technology for Economic and Clinical Health Act (“HITECH”), and their respective implementing
regulations, which imposes privacy, security, and breach reporting obligations with respect to Protected Health Information (“PHI”),
upon entities subject to the law, such as health plans, healthcare clearinghouses and certain healthcare providers, and their respective
business associates that perform services on their behalf that involve PHI. HITECH also created new tiers of civil monetary penalties,
amended HIPAA to make HIPAA compliance as well as civil and criminal penalties directly applicable to business associates, and gave
state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the HIPAA laws
and seek attorneys’ fees and costs associated with pursuing federal civil actions; and |
| ● |
analogous
state and foreign law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply
to items or services reimbursed by any third-party payer, including commercial insurers or patients; state laws that require device
companies to comply with the industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated
by the federal government or otherwise restrict payments that may be made to healthcare providers and other potential referral sources;
state and local laws that require the licensure of sales representatives; state laws that require device manufacturers to report
information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures
and pricing information; data privacy and security laws and regulations in foreign jurisdictions that may be more stringent than
those in the United States (such as the EU, which adopted the General Data Protection Regulation, which became effective in May 2018);
state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other
in significant ways and may not have the same effect, thus complicating compliance efforts; and state laws related to insurance fraud
in the case of claims involving private insurers. |
These
laws and regulations, among other things, constrain our business, marketing, and other promotional activities by limiting the kinds of
financial arrangements, including sales programs, we may have with physicians or other potential purchasers of our products. We have
also entered into consulting agreements with physicians, which are subject to these laws. Further, while we do not submit claims and
our future customers will make the ultimate decision on how to submit claims, we may provide reimbursement guidance and support regarding
our products. Due to the breadth of these laws, the narrowness of statutory exceptions and regulatory safe harbors available, and the
range of interpretations to which they are subject, it is possible that some of our current or future practices might be challenged under
one or more of these laws.
Certain
enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which
has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. For example, U.S. federal
and state regulatory and enforcement agencies continue to actively investigate violations of healthcare laws and regulations, including
pursuing novel theories of liability under these laws. These government agencies recently have increased regulatory scrutiny and enforcement
activity with respect to manufacturer reimbursement support activities and patient support programs, including bringing criminal charges
or civil enforcement actions under the federal healthcare Anti-Kickback statute, federal civil False Claims Act, the health care fraud
statute, and HIPAA privacy provisions. Responding to investigations can be time and resource consuming and can divert management’s
attention from the business. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our
business. Even an unsuccessful challenge or investigation into our practices could cause adverse publicity, and be costly to respond
to.
If
our operations are found to be in violation of any of the healthcare laws or regulations described above or any other healthcare regulations
that apply to us, we may be subject to administrative, civil and criminal penalties, damages, fines, disgorgement, substantial monetary
penalties, exclusion from participation in government healthcare programs, such as Medicare and Medicaid, imprisonment, additional reporting
obligations, and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance
with these laws, reputational harm, and the curtailment or restructuring of our operations.
Our
products may cause or contribute to adverse medical events that we are required to report to the FDA and other governmental authorities,
and if we fail to do so, we would be subject to sanctions that could harm our reputation, business, results of operations, and financial
condition. The discovery of serious safety issues with our products, or a recall of our products either voluntarily or at the direction
of the FDA or another governmental authority, could have a negative impact on us.
We
are required to timely file various reports with the FDA, including reports required by the medical device reporting regulations which
require us to report to the FDA when we receive or become aware of information that reasonably suggests that one of our products may
have caused or contributed to a death or serious injury or malfunctioned in a way that, if the malfunction were to recur in the device
or a similar device that we market, could cause or contribute to a death or serious injury. If we fail to comply with our reporting obligations,
the FDA or other governmental authorities could take action, including warning letters, untitled letters, administrative actions, criminal
prosecution, imposition of civil monetary penalties, revocation of our device clearance, seizure of our products, or delay in clearance
of future products. The FDA and certain foreign regulatory bodies have the authority to require the recall of commercialized products
under certain circumstances.
A
government-mandated or voluntary recall by us could occur as a result of an unacceptable risk to health, component failures, malfunctions,
labeling or design deficiencies, packaging defects, or other deficiencies, or failures to comply with applicable regulations. If we do
not adequately address problems associated with our devices, we may face additional regulatory requirements or enforcement action, including
required new marketing authorizations, FDA warning letters, product seizure, injunctions, administrative penalties, or civil or criminal
proceedings.
We
may initiate voluntary withdrawals, removals, or corrections for our products in the future that we determine do not require notification
of the FDA because no material compliance issue or safety risk is involved. If the FDA disagrees with our determinations, it could require
us to report those actions and we may be subject to enforcement action. A future recall announcement or other corrective action could
harm our financial results and reputation, potentially lead to product liability claims against us, require the dedication of our time
and capital, and negatively affect our sales.
In
addition, the FDA’s and other regulatory authorities’ policies may change, and additional government regulations may be enacted
that could prevent, limit, or delay regulatory clearance or approval of our future products. For example, in November 2018, the FDA announced
that it plans to develop proposals to drive manufacturers utilizing the 510(k) pathway toward the use of newer predicates. It is unclear
the extent to which any proposals, if adopted, could impose additional regulatory requirements on us that could delay our ability to
obtain new 510(k) clearances, increase the costs of compliance, or restrict our ability to maintain our current clearances.
We
also cannot predict the likelihood, nature, or extent of government regulation that may arise from future legislation or administrative
or executive action, either in the U.S. or abroad. For example, the Trump Administration previously enacted several executive actions
that could impose significant burdens on, or otherwise materially delay, the FDA’s ability to engage in routine regulatory and
oversight activities. It is difficult to predict how these executive actions and executive actions that may be taken under the Biden
Administration or future administrations may affect the FDA’s ability to exercise its regulatory authority. If these executive
actions impose constraints on the FDA’s ability to engage in oversight and implementation activities in the normal course, our
business may be negatively impacted.
Disruptions
at the FDA, other agencies or notified bodies caused by funding shortages or global health concerns could hinder their ability to hire,
retain, or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, cleared or
approved, or commercialized in a timely manner, or at all, which could negatively impact our business.
The
ability of the FDA, other agencies and notified bodies to review and authorize or certify for marketing new products can be affected
by a variety of factors, including government budget and funding levels, statutory, regulatory and policy changes, agency’s or
notified body’s ability to hire and retain key personnel and accept the payment of user fees, and other events that may otherwise
affect the agency’s or notified body’s ability to perform routine functions. Average review times at the FDA and other agencies
and notified bodies have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund
research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at
the FDA, other agencies and notified bodies may also slow the time necessary for new medical devices or modifications to be reviewed
and/or cleared, approved or certified by necessary agencies or notified bodies, which would adversely affect our business. For example,
over the last several years, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had
to furlough critical FDA employees and stop critical activities. If a prolonged government shutdown occurs, or if global health concerns,
including pandemics, were to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other
regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process
our regulatory submissions, which could have a material adverse effect on our business.
In
the EU, notified bodies must be officially designated to certify products and services in accordance with the MDR, which regulates the
development and sale of medical devices in Europe. While several notified bodies have been designated, the COVID-19 pandemic significantly
slowed down their designation process and the current designated notified bodies are facing a large amount of requests with the new regulation,
as a consequence of which review times have lengthened although a regulation amending the EU MDR was adopted in March 2023, extending
existing transitional provisions to December 31, 2028. Despite a recent increase in designations, the current number of notified bodies
designated under the MDR remains significantly lower than the number of notified bodies designated under the previous regime. The current
designated notified bodies are therefore facing a backlog of requests as a consequence of which review times have lengthened. This situation
could impact our ability to grow our business in the EU and EEA and the ability of the notified body to timely review and process our
regulatory submissions and perform its audits.
Changes
in internet regulations could adversely affect our business.
Laws,
rules, and regulations governing internet communications, advertising, and e-commerce are dynamic, and the extent of future government
regulation is uncertain. Federal and state regulations govern various aspects of our online business, including intellectual property
ownership and infringement, trade secrets, the distribution of electronic communications, marketing and advertising, user privacy and
data security, search engines, and internet tracking technologies. Future taxation on the use of the internet or e-commerce transactions
could also be imposed. Existing or future regulation or taxation could increase our operating expenses and expose us to significant liabilities.
Increased
focus on current and anticipated environmental, social and governance (“ESG”) laws and increased
scrutiny of our ESG policies and practices may materially increase our costs, expose us to potential liability, adversely impact our
reputation, employee retention, willingness of potential customers and suppliers to do business with us and willingness of investors
to invest in us.
Our
operations are subject to a variety of existing local, regional and global ESG laws and regulations, and we will likely be required to
comply with new, broader, more complex and more costly laws and regulations that focus on ESG matters. Our compliance obligations will
likely span all aspects of our business and operations, including product design and development, materials sourcing and other procurement
activities, product packaging, product safety, energy and natural resources usage, facilities design and utilization, recycling and collection,
transportation, disposal activities and workers’ rights.
Environmental
regulations related to greenhouse gases are expected to have an increasingly larger impact on our or our suppliers’ energy sources.
Many U.S. and foreign regulators have enacted or are considering enacting new or additional disclosure requirements or limits on the
emissions of greenhouse gases, including, but not limited to, carbon dioxide and methane, from power generation units using fossil fuels.
The effects of greenhouse gas emission limits on power generation are subject to significant uncertainties, including the timing of any
new requirements, levels of emissions reductions and the scope and types of emissions regulated. These limits may have the effect of
increasing our costs and those of our suppliers and could result in manufacturing, transportation and supply chain disruptions and delays
if clean energy alternatives are not readily available in adequate amounts when required. Moreover, alternative energy sources, coupled
with reduced investments in traditional energy sources and infrastructure, may fail to provide the predictable, reliable, and consistent
energy that we, our suppliers and other businesses need for operations.
Meeting
our obligations under existing ESG laws, rules, or regulations is already costly to us and our suppliers, and we expect those costs to
increase as new laws are enacted, possibly materially. Additionally, we expect regulators to perform investigations, inspections and
periodically audit our compliance with these laws and regulations, and we cannot provide assurance that our efforts or operations will
be compliant. If we fail to comply with any requirements, we could be subject to significant penalties or liabilities and we may be required
to implement new and materially more costly processes and procedures to come into compliance. Further, these laws are subject to unpredictable
changes. Even if we successfully comply with these laws and regulations, our suppliers may fail to comply. We may also suffer financial
and reputational harm if future customers require, and we are unable to deliver, certification that our products are conflict free. In
all of these situations, our future customers may stop purchasing products from us, and may take legal action against us, which could
harm our reputation, revenues and results of operations.
Investor
advocacy groups, institutional investors, investment funds, proxy advisory services, stockholders, and consumers are also increasingly
focused on corporate ESG practices. Additionally, public interest and legislative pressure related to public companies’ ESG practices
continues to grow. If our ESG practices fail to meet investors’ or other industry stakeholders’ evolving expectations and
standards, including environmental stewardship, support for local communities, board of director and employee diversity, human capital
management, employee health and safety practices, product quality, supply chain management, corporate governance and transparency and
employing ESG strategies in our operations, our brand, reputation and employee retention may be negatively impacted, potential customers
and suppliers may be unwilling to do business with us and investors may be unwilling to invest in us. In addition, as we work to align
our ESG practices with industry standards, we have expanded and will likely continue to expand our disclosures in these areas. We also
expect to incur additional costs and require additional resources to monitor, report, and comply with our various ESG practices. If we
fail to adopt ESG standards or practices as quickly as stakeholders desire, report on our ESG efforts or practices accurately, or satisfy
the disclosure and other expectations of stakeholders, our reputation, business, financial performance, growth, and stock price may be
adversely impacted.
We
are subject to consumer protection laws that regulate our marketing practices and prohibit unfair or deceptive acts or practices. Our
actual or perceived failure to comply with such obligations could harm our business, and changes in such regulations or laws could require
us to modify our products, marketing or advertising efforts.
In
connection with the marketing or advertisement of our products and services, we could be the target of claims relating to false, misleading,
deceptive, or otherwise noncompliant advertising or marketing practices, including under the auspices of the FTC and state consumer protection
statutes. If we rely on third parties to provide any marketing and advertising of our products and services, we could be liable for,
or face reputational harm as a result of, their marketing practices if, for example, they fail to comply with applicable statutory and
regulatory requirements.
If
we are found to have breached any consumer protection, advertising, unfair competition, or other laws or regulations, we may be subject
to enforcement actions that require us to change our marketing and business practices in a manner which may negatively impact us. This
could also result in litigation, fines, penalties, and adverse publicity that could cause reputational harm and loss of patient trust,
which could have an adverse effect on our business.
Risks
Related to Our Operations in Israel
We
conduct our operations in Israel. Conditions in Israel, including the recent attack by Hamas and other terrorist organizations from the
Gaza Strip, and the region, and Israel’s war against them, may affect our operations.
We
have operations in the State of Israel, and we are directly affected by political, economic and security conditions in that country.
Since the establishment of the State of Israel in 1948, a number of armed conflicts have taken place between Israel and its Arab neighbors
and a state of hostility, varying in degree and intensity, has led to security and economic problems for Israel. In addition, acts of
terrorism, armed conflicts or political instability in the region could negatively affect local business conditions and harm our results
of operations. We cannot predict the effect on the region of any diplomatic initiatives or political developments involving Israel or
the Palestinians or other countries and territories in the Middle East. Recent political events, including political uprisings, social
unrest and regime change, in various countries in the Middle East and North Africa have weakened the stability of those countries and
territories, which could result in extremists coming to power. In addition, Iran has threatened to attack Israel and is widely believed
to be developing nuclear weapons. Iran is also believed to have a strong influence among extremist groups in the region, such as Hamas
in Gaza and Hezbollah in Lebanon. This situation has escalated in the past and may potentially escalate in the future to violent events
which may affect Israel and us. On October 7, 2023, the State of Israel was attacked by and subsequently declared war on Hamas. Israel
has been in an ongoing state of war with Hamas since that time. Following the attack by Hamas, Hezbollah, a terrorist organization in
Lebanon has also launched missile, rocket, and shooting attacks against Israeli military sites, troops, and Israeli towns in northern
Israel. In response to these attacks, the Israeli army has carried out a number of targeted strikes on sites belonging to Hezbollah in
southern Lebanon and in October 2024, the Israeli military initiated a ground operation in Lebanon, primarily near the Israel-Lebanon
border. In November 2024, Israel entered into a ceasefire agreement with Hezbollah, but there are no guarantees as to whether the agreement
will hold or whether further hostilities will resume.
During
2024, Iran launched missile and unmanned aerial vehicle, or UAV, attacks on Israel. Most of the missiles and UAVs were intercepted by
Israel’s defense systems, with support from the United States and other countries, including regional allies, preventing significant
damage and resulting in no casualties. Despite the successful interceptions, the attacks posed an elevated threat to Israel’s security.
On June 13, 2025, in light of continued nuclear threats and intelligence assessments indicating imminent attacks, Israel launched a preemptive
strike directly targeting military and nuclear infrastructure inside Iran aimed to disrupt Iran’s capacity to coordinate or launch
further hostilities against Israel, as well as disrupt its nuclear program. For 12 days, both sides launched attacks against one another,
with Iran targeting civilian infrastructure. As a result of the escalation with Iran, Israel temporarily closed its airspace and ceased
all port activity related to commercial shipments. On June 22, 2025, the U.S. military joined Israel in launching strikes directly targeting
nuclear infrastructure in Iran. A U.S. brokered ceasefire took effect on June 24, 2025.
On
February 28, 2026, Israel and the United States commenced coordinated military strikes against targets in Iran, including military and
strategic infrastructure in response to ongoing regional tensions and recent escalations involving Iran’s nuclear and military
activities. In response, Iran launched a series of retaliatory attacks against Israel, targeting major cities and strategic sites, which
are ongoing. While most of these attacks have been intercepted to date, some resulted in civilian casualties and damage to property.
Subsequently, Hezbollah launched attacks against Israel in retaliation for the killing of Ali Hosseini Khamenei, the former Supreme Leader
of Iran, and in response, Israel launched attacks against Lebanon and Israeli ground forces have entered into Southern Lebanon, and hostilities
between Israel and Hezbollah are ongoing. Iran subsequently began launching retaliatory strikes against U.S. and other targets in the
Gulf region. The Israeli government has raised its alert level nationwide, and the situation remains highly unstable, with ongoing exchanges
of fire and heightened risk of further escalation. Regional and international responses are ongoing, and the risk of broader conflict
in the Middle East has increased.
In
addition, in December 2024, Ba’athist Syria, led by President Bashar al-Assad, collapsed during a major offensive by opposition
forces made up of several competing rebel groups. In response, the Israeli Defense Forces took control over a United Nations-designated
buffer zone over Mount Hermon that separates Israel and Syria. Simultaneously, Israel conducted targeted military strikes against military
assets in Syria, aiming to eliminate any chemical weapons storage sites that could be used by rebel groups and further weaken Iran’s
operational capabilities in the region. While the transitional government of Syria has indicated that it is interested in reconstruction
and stability rather than a continuation of conflicts with Israel, there are no guarantees that there will be no future escalation of
hostilities or that Syria will not permit other neighboring countries to launch attacks at Israel from its territory.
Any
hostilities involving Israel, or the interruption or curtailment of trade within Israel or between Israel and its trading partners, or
the ability to ship our products overseas, could adversely affect our operations and results of operations and could make it more difficult
for us to raise capital. Parties with whom we may do business have sometimes declined to travel to Israel during periods of heightened
unrest or tension, forcing us to make alternative arrangements when necessary. The conflict situation in Israel could cause situations
where medical product certifying or auditing bodies could not be able to visit manufacturing facilities of our subcontractors in Israel
in order to review our certifications or clearances, thus possibly leading to temporary suspensions or even cancellations of our product
clearances or certifications. The conflict situation in Israel could also result in parties with whom we have agreements involving performance
in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions
in such agreements.
There
have been travel advisories issued related to travel to Israel, restriction on travel, and delays and disruptions as related to imports
and exports may be imposed in the future. An inability to receive supplies and materials, shortages of materials or difficulties in procuring
our materials, among others, or conversely, our ability to ship products to our US facilities or overseas customers, may adversely impact
our ability to commercialize and manufacture our product candidates and products in a timely manner. This could cause a number of delays
and/or issues for our operations, including delay of the review of our product candidates by regulatory agencies, which in turn would
have a material adverse impact on our ability to commercialize our product candidates.
We
currently have 12 full-time employees, including 5 employees who are members of senior management, as well as engagements with 2 contractors,
who are located in and/or reside in Israel. As a result, our business and operations are directly affected by economic, political,
geopolitical and military conditions in Israel. Shelter-in-place and work-from-home measures, government-imposed restrictions on movement
and travel, and other precautions taken to address the ongoing conflict may temporarily disrupt our management and employees’ ability
to effectively perform their daily tasks.
The
IDF, the national military of Israel, is a conscripted military service, subject to certain exceptions. None of our employees are subject
to military service in the IDF and have been called to serve, but many do serve on guard duty in their local communities from time to
time. It is possible that there will be further military reserve duty call-ups in the future, which may affect our business due to a
shortage of skilled labor and loss of institutional knowledge, and necessary mitigation measures we may take to respond to a decrease
in labor availability, such as overtime and third-party outsourcing, for example, which may have unintended negative effects and adversely
impact our results of operations, liquidity, or cash flows.
It
is currently not possible to predict the duration or severity of the ongoing conflict or its effects on our business, operations, and
financial conditions. The ongoing conflict is rapidly evolving and developing, and could disrupt our business and operations, interrupt
our sources and availability of supply, and hamper our ability to raise additional funds or sell our securities, among others.
The
impact of planned changes in the Israeli Judicial System on capital raising in the high-tech sector is difficult to predict.
In
January and February 2023, the Israeli government began promoting a plan to implement changes in the judicial system in Israel, as well
as additional legislative changes. According to various assessments and publications, the proposed changes (some of which have already
passed the first, second, and even third readings in the Knesset) are causing significant controversy and, therefore, may also impact
the performance and resilience of the Israeli economy. According to some forecasts, this plan may lead, among other things, to a downgrade
in Israel’s credit rating, damage to the local currency, increased inflation, a reduction in investments in the Israeli economy,
capital outflow from Israel, an increase in the cost of capital raising in the Israeli economy, and harm to the activity of the economic
sector in general and the high-tech sector in particular. The forecast from the Bank of Israel’s research unit in July 2023 provided
evidence of a decline in the volume of fundraising for investments in start-up companies in Israel.
Since
October 2023, following the start of Israel-Hamas war, public and media focus on legislative changes has diminished. It is currently
not possible to predict whether the legislative efforts will be renewed and or their effects on our business, operations and financial
conditions.
Our
“Israeli identity” may have negative impact on our sales.
Part
of our management, and the majority of development, are based in Israel, while all our product sales including operations are made outside
of Israel. Accordingly, the political status of the State of Israel may impact our activity. The Israeli identity sometimes serves as
a sales promoter (due to the recognition of Israel’s technological advantages), while in other cases, it may be a disadvantage
and could even lead to the cancellation of deals (such as within the framework of coordinated efforts to boycott Israeli products and/or
divest from Israel). Additionally, some countries worldwide have imposed or may impose restrictions on doing business in or with Israeli
companies from time to time.
Because
a certain portion of our expenses is incurred in currencies other than the U.S. dollar, our results of operations may be harmed by currency
fluctuations and inflation.
We
expect our revenues from future licensing agreements to be denominated mainly in U.S. dollars or in Euros. We pay a substantial portion
of our expenses in U.S. dollars; however, a portion of our expenses, related to salaries of the employees in Israel and payment to part
of the service providers in Israel and other territories, are paid in New Israeli Shekels, or NIS, and in other currencies. In addition,
a portion of our financial assets is held in NIS and in other currencies. As a result, we are exposed to the currency fluctuation risks,
and we do not attempt to hedge against such risks. For example, if the NIS strengthens against the U.S. dollar, our reported expenses
in U.S. dollars may be higher than anticipated. In addition, if the NIS weakens against the U.S. dollar, the U.S. dollar value of our
financial assets held in NIS will decline.
It
may be difficult for investors in the United States to enforce any judgments obtained against us or any of our directors or officers.
Almost
all of our assets are located outside the United States, although we do maintain a permanent place of business within the United States.
In addition, some of our officers and directors are nationals and/or residents of countries other than the United States, and all or
a substantial portion of such persons’ assets are located outside the United States. As a result, it may be difficult for investors
to enforce within the United States any judgments obtained against us or any of our non-U.S. directors or officers, including judgments
predicated upon the civil liability provisions of the securities laws of the United States or any state thereof. Additionally, it may
be difficult to assert U.S. securities law claims in actions originally instituted outside of the United States. Israeli courts may refuse
to hear a U.S. securities law claim because Israeli courts may not be the most appropriate forums in which to bring such a claim. Even
if an Israeli court agrees to hear a claim, it may determine that the Israeli law, and not U.S. law, is applicable to the claim.
Further,
if U.S. law is found to be applicable, certain content of applicable U.S. law must be proved as a fact, which can be a time-consuming
and costly process, and certain matters of procedure would still be governed by the Israeli law. Consequently, you may be effectively
prevented from pursuing remedies under U.S. federal and state securities laws against us or any of our non-U.S. directors or officers.
Risks
Related to Our Intellectual Property
Our
success depends in part on our proprietary technology, and if we are unable to successfully enforce our intellectual property rights,
our competitive position may be harmed.
Our
success will depend in part on our ability to maintain existing intellectual property and to obtain and maintain further intellectual
property protection for our products and services, both in the U.S. and in other countries. We intend to protect our intellectual property
rights, including our AI technology and related algorithms, through a combination of patent, trademark, copyright, and trade secret laws,
as well as third-party confidentiality and assignment agreements. Our inability to do so could harm our competitive position.
We
rely on our portfolio of issued and pending patent applications in the U.S. and other countries to protect a large part of our intellectual
property and our competitive position; however, our currently pending or future patent filings may not result in the issuance of patents.
While we generally apply for patents in those countries where we intend to make, have made, use, or sell patented products, we may not
accurately predict all of the countries where patent protection will ultimately be desirable. If we fail to timely file for a patent,
we may be precluded from doing so at a later date.
Patent
rights are territorial, and patent protection extends only to those countries where we have issued patents. Filing, prosecuting and defending
patents on our products and our future products in all countries and jurisdictions throughout the world would be prohibitively expensive,
and our intellectual property rights in some countries outside the United States could be less extensive than those in the United States.
Many countries do not protect intellectual property to the same extent as the U.S. or Europe, and their litigation processes differ.
Competitors may successfully challenge or avoid our patents, or manufacture products in countries where we have not applied for patent
protection. Changes in the patent laws in the U.S. or other countries may diminish the value of our patent rights. As a result of these
and other factors, the scope, validity, enforceability, and commercial value of our patent rights are uncertain and unpredictable.
Furthermore,
the patent positions of medical device companies involve complex legal and factual questions, and, therefore, the issuance, scope, validity
and enforceability of any patent claims that we may obtain cannot be predicted with certainty. The issuance of a patent, while presumed
valid and enforceable, is not conclusive as to its validity or its enforceability and it may not provide us with adequate proprietary
protection or competitive advantages against competitors with similar products. Any patents issued to us may be challenged, invalidated,
held unenforceable, circumvented, or may not be sufficiently broad to prevent third parties from producing competing products similar
in design to our products. In addition, any protection afforded by foreign patents may be more limited than that provided under U.S.
patent and intellectual property laws. There can be no assurance that any of our patents, any patents licensed to us, or any patents
which we may be issued in the future, will provide us with a competitive advantage or afford us protection against infringement by others,
or that the patents will not be successfully challenged or circumvented by third parties, including our competitors. Further, there can
be no assurance that we will have adequate resources to enforce our patents. Competitors may also be able to design around our patents.
Other parties may develop and obtain patent protection for more effective technologies, designs or methods.
Our
ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise
the components that are used in their products. Moreover, it may be difficult or impossible to obtain evidence of infringement in a competitor’s
or potential competitor’s product, particularly in litigation in countries other than the U.S. that do not provide an extensive
discovery procedure. Any litigation to enforce or defend our patent rights, if any, even if we were to prevail, could be costly and time-consuming
and would divert the attention of our management and key personnel from our business operations. We may not prevail in any lawsuits that
we initiate and the damages or other remedies awarded if we were to prevail may not be commercially meaningful.
Moreover,
advances in AI technology may generate developments that existing IP laws do not adequately protect. The legislative and regulatory environment
is out of our control, may change rapidly and unpredictably, and may negatively influence our revenue, costs, earnings, and growth. Some
rules and regulations may be subject to litigation or other challenges that delay or modify their implementation and impact on us.
We
also may seek to rely on protection of copyright, trade secrets, know-how, and confidential and proprietary information. We generally
enter into confidentiality and non-compete agreements with our employees, consultants, and collaborative partners upon their commencement
of a relationship with us. However, these agreements may not provide meaningful protection against the unauthorized use or disclosure
of our trade secrets or other confidential information, and adequate remedies may not exist if unauthorized use or disclosure were to
occur. The exposure of our trade secrets and other proprietary information would impair our competitive advantages and could have a material
adverse effect on our operating results, financial condition, and future growth prospects. In particular, a failure to protect our proprietary
rights might allow competitors to copy our technology, which could adversely affect our pricing and market share. We may not be able
to prevent the unauthorized disclosure or use of our technical knowledge or trade secrets by consultants, vendors, former employees and
current employees. Further, other parties may independently develop substantially equivalent know-how and technology.
We
currently own registered trademarks for our products, and we intend to rely on both registered and common law rights for our trademarks
in the future. There can be no assurance that our future trademark applications will be approved. Third parties may also oppose our trademark
applications, or otherwise challenge our use of the trademarks. In the event that our trademarks are successfully challenged, we could
be forced to rebrand our products and services, which could result in loss of brand recognition, and could require us to devote resources
to advertising and marketing new brands. Further, there can be no assurance that competitors will not infringe our trademarks, or that
we will have adequate resources to enforce our trademarks.
Litigation,
interferences, oppositions, re-exams, inter partes reviews, post grant reviews, or other proceedings are, have been, and may in the future
be necessary in some instances to determine the validity and scope of certain of our proprietary rights, and in other instances to determine
the validity, scope, or non-infringement of certain proprietary rights claimed by third parties to be pertinent to the manufacture, use,
or sale of our products or provision of our services. These types of proceedings are unpredictable and may be protracted, expensive,
and distracting to management. The outcome of such proceedings could adversely affect the validity and scope of our patent or other proprietary
rights, hinder our ability to manufacture and market our products and provide our services, require us to seek a license for the infringed
product or technology, or result in the assessment of significant monetary damages. An unfavorable ruling could include monetary damages
or, in cases where injunctive relief is sought, an injunction prohibiting us from selling our products or providing our services. Any
of these results from litigation could adversely affect our business, financial condition, and results of operations.
Successful
cybersecurity attacks, data breaches, unapproved use of machine learning or AI tools, or other security incidents could result in the
loss of IP and key technological advantages. Security incidents could result in, for example, unauthorized access to, disclosure, modification,
misuse, loss, or destruction of company, patient, or other third party data; theft or import of sensitive, regulated, or confidential
data including personal information and IP, such as key innovations in AI; the loss of access to critical data or systems through ransomware;
and business delays.
If
we infringe or violate the patents or proprietary rights of other parties or are subject to an intellectual property infringement or
misappropriation claim, our ability to grow our business may be severely limited.
Our
commercial success also depends upon our ability, and the ability of any third party with which we may partner, to develop, manufacture,
market and sell our products, if approved, and use our patent-protected technologies without infringing the patents of third parties.
Extensive litigation over patents and other intellectual property rights is common in the medical device industry.
We
may not have identified all patents, published applications or published literature that affect our business either by blocking our ability
to commercialize our products, by preventing the patentability of one or more aspects of our products, or by covering the same or similar
technologies that may affect our ability to market our products. For example, we may not have conducted a patent clearance search sufficient
to identify potentially obstructing third party patent rights. Moreover, patent applications in the United States are maintained in confidence
for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the U.S. Patent and Trademark
Office, or the USPTO, for the entire time prior to issuance as a U.S. patent. Patent applications filed in countries outside of the United
States are not typically published until at least 18 months from their first filing date. Similarly, publication of discoveries in scientific
or patent literature often lags behind actual discoveries. We cannot be certain that we were the first to invent, or the first to file,
patent applications covering our products. We also may not know if our competitors filed patent applications for technology covered by
our pending applications or if we were the first to invent the technology that is the subject of our patent applications. Competitors
may have filed patent applications or received patents and may obtain additional patents and proprietary rights that block or compete
with our patents.
We
may therefore in the future be the subject of patent or other litigation. From time to time, we may in the future receive letters from
third parties drawing our attention to their patent rights. While we do not believe that we infringe upon any valid and enforceable rights
that have been brought to our attention, and we take necessary steps to ensure that we do not infringe on the rights of others, there
may be other more pertinent rights of which we are presently unaware. The defense and prosecution of intellectual property suits, interference
proceedings, and related legal and administrative proceedings could result in substantial expense to us and significant diversion of
effort by our technical and management personnel. An adverse determination of any litigation or interference proceeding to which we may
become a party could subject us to significant liabilities. An adverse determination of this nature could also put our patents at risk
of being invalidated or interpreted narrowly or require us to seek licenses from third parties. Licenses may not be available on commercially
reasonable terms or at all, in which event, our business would be materially adversely affected. Intellectual property litigation or
claims could force us to cease developing, selling or otherwise commercializing one or more of our products; to pay substantial damages
for past use of the asserted intellectual property; and redesign, or rename in the case of trademark claims, our product(s) to avoid
such third party rights, which may not be possible or which could be costly and time-consuming. Any of these risks coming to fruition
could have a material adverse effect on our business, results of operations, financial condition and prospects.
Our
failure to secure trademark registrations could adversely affect our ability to market our products and operate our business.
Any
future trademark applications in the United States and any other jurisdictions where we may file may not be allowed registration, and
we may not be able to maintain or enforce our registered trademarks. During trademark registration proceedings, we may receive rejections.
Although we are given an opportunity to respond to those rejections, we may be unable to overcome such rejections. In addition, in the
USPTO and in corresponding foreign agencies, third parties are given an opportunity to oppose pending trademark applications and to seek
to cancel registered trademarks. Opposition or cancellation proceedings may be filed against our applications and/or registrations, and
our applications and/or registrations may not survive such proceedings. Failure to secure such trademark registrations in the United
States and in foreign jurisdictions could adversely affect our ability to market our products and our business.
We
may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As
is common in the medical device industry, we may employ individuals who were previously employed at other companies similar to ours,
including our competitors or potential competitors. We may become subject to claims that these employees or we have inadvertently or
otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to
defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs
and be a distraction to management.
Obtaining
and maintaining patent protection depends on compliance with various procedures and other requirements, and our patent protection could
be reduced or eliminated in case of non-compliance with these requirements.
Periodic
maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and/or applications will be due to the relevant
patent agencies in several stages over the lifetime of the patents and /or applications. The relevant patent agencies require compliance
with a number of procedures, documentation, fee payment and other provisions during the patent application process. In many cases, an
inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. However, there are
situations in which the failure to comply with the relevant requirements can result in the abandonment or lapse of the patent or patent
application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. In such an event, our competitors might
be able to use our technologies and know-how which could have a material adverse effect on our business, prospects, financial condition
and results of operation.
Patent
terms may be inadequate to protect our competitive position on our products for an adequate amount of time.
Patents
have a limited lifespan. In the United States, if all maintenance fees are timely paid, the natural expiration of a patent is generally
20 years from its earliest U.S. non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection
it affords, is limited. Even if patents covering our products are obtained, once the patent life has expired for a product, we may be
open to competition from competitive products. Given the amount of time required for the development, testing and regulatory review of
new products, patents protecting such products might expire before or shortly after such products are commercialized. As a result, our
patent portfolio may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We
may be subject to claims challenging the inventorship of our patents and other intellectual property.
We
may be subject to claims that former employees, collaborators or other third parties have an interest in our patents or other intellectual
property as an inventor or co-inventor or an author. For example, we may have inventorship or ownership disputes arise from conflicting
obligations of consultants or others who are involved in developing our products. Litigation may be necessary to defend against these
and other claims challenging inventorship or our ownership of our patents or other intellectual property. If we fail in defending any
such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of,
or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are
successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other
employees.
We
use AI in our business, and challenges with properly managing its use could result in reputational harm, competitive harm, and legal
liability, and adversely affect our results of operations.
We
incorporate AI solutions into our ENvue System, services, and features, and these applications are important in our operations. Our competitors
or other third parties may incorporate AI into their products more quickly or more successfully than us, which could impair our ability
to compete effectively and adversely affect our results of operations.
Additionally,
if the content, analyses, or recommendations that AI applications assist in producing are or are alleged to be deficient, inaccurate,
or biased, our business, financial condition, and results of operations may be adversely affected. Our use of AI and machine learning
is subject to risks related to flaws in our algorithms and datasets that may be insufficient or contain biased information. The development
of AI technologies is complex, and there are several challenges associated with achieving the desired level of accuracy, efficiency,
and reliability. The algorithms and models used in our AI systems may have limitations, including biases, errors, or inability to handle
certain data types or scenarios. There is a risk of system failures, disruptions, or vulnerabilities that could compromise the integrity,
security, or privacy of our platform. These failures could result in reputational damage, legal liabilities, or loss of user confidence,
which could materially affect our business.
The
use of AI applications has resulted in, and may in the future result in, cybersecurity incidents that implicate the personal data of
patients and users of such applications. Any such cybersecurity incidents related to our use of AI applications could adversely affect
our reputation and results of operations. AI also presents emerging ethical issues, and if our use of AI becomes controversial, we may
experience brand or reputational harm, competitive harm, or legal liability. The rapid evolution of AI, including potential government
regulation of AI, will require significant resources to develop, test and maintain our platform, services, and features to help us implement
AI ethically in order to minimize unintended, harmful impact.
Legislative
and governmental activity in the privacy area may result in new laws or regulations that are applicable to us and that may hinder our
business, for example, by restricting use or sharing of patient data, limiting our ability to provide certain data to our customers,
limiting our ability to develop or modify our AI systems, or otherwise regulating AI and machine learning, including the use of algorithms
and automated processing in ways that could materially affect our business, or which may lead to significant increases in the cost of
compliance.
Risks
Related to Our Organization and Securities
The
price of our securities may be volatile, and the market price of our securities may drop below the price you pay.
We
expect that the price of our securities will fluctuate significantly. Market prices for securities of early-stage medical device companies
have historically been particularly volatile. In addition to the factors discussed in this “Risk Factors” section and elsewhere
in this Annual Report, these factors include:
| ● |
progress,
or lack of progress, in developing and commercializing our products; |
| ● |
favorable
or unfavorable decisions about our products or intellectual property from government regulators, insurance companies or other third-party
payers; |
| ● |
our
ability to recruit and retain qualified regulatory and research and development personnel; |
| ● |
changes
in investors’ and securities analysts’ perception of the business risks and conditions of our business; |
| ● |
changes
in our relationship with key collaborators; |
| ● |
changes
in the market valuation or earnings of our competitors or companies viewed as similar to us; |
| ● |
changes
in key personnel; |
| ● |
depth
of the trading market in our common stock; |
| ● |
changes
in our capital structure, such as future issuances of securities or the incurrence of additional debt; |
| ● |
the
granting or exercise of employee stock options or other equity awards; |
| ● |
realization
of any of the risks described under this section entitled “Risk Factors”; and |
| ● |
general
market and economic conditions. |
In
recent years, the stock markets, in general, have experienced extreme price and volume fluctuations especially in the biotechnology sector.
Broad market and industry factors may materially harm the market price of shares of our common stock. In the past, following periods
of volatility in the market price of a company’s securities, securities class action litigation has often been instituted against
that company. If we were involved in any similar litigation, we could incur substantial costs and our management’s attention and
resources could be diverted. In the recent past, the U.S. and global markets have been experiencing volatility and disruption following
the escalation of geopolitical tensions and the start of the military conflict between Russia and Ukraine, and Israel and certain hostile
entities. A continuation or worsening of the levels of market disruption and volatility could have an adverse effect on our ability to
access capital, on our business, results of operations and financial condition, and on the market price of our common stock.
We
have a significant number of warrants and options, and future sales of our common stock upon exercise of these options or warrants, or
the perception that future sales may occur, may cause the market price of our common stock to decline, even if our business is doing
well.
Sales
of a significant number of shares of our common stock in the public market could harm the market price of our common stock and make it
more difficult for us to raise funds through future offerings of common stock. Our stockholders and the holders of our outstanding warrants
and options, upon exercise of these options or warrants, may sell substantial amounts of our common stock in the public market. The availability
of these shares of our common stock for resale in the public market has the potential to cause the supply of our common stock to exceed
investor demand, thereby decreasing the price of our common stock.
In
addition, the fact that our stockholders and holders of our warrants and options can sell substantial amounts of our common stock in
the public market, whether or not sales have occurred or are occurring, could make it more difficult for us to raise additional financing
through the sale of equity or equity-related securities in the future at a time and price that we deem reasonable or appropriate.
If
we fail to comply with the continued listing requirements of Nasdaq, our common stock may be delisted and the price of our common stock
and our ability to access the capital markets could be negatively impacted.
Our
common stock is currently listed for trading on Nasdaq. We must satisfy Nasdaq’s continued listing requirements, including, among
other things, a minimum stockholders’ equity of $2.5 million and a minimum closing bid price of $1.00 per share or risk
delisting, which would have a material adverse effect on our business. A delisting of our common stock from Nasdaq could materially reduce
the liquidity of our common stock and result in a corresponding material reduction in the price of our common stock. In addition, delisting
could harm our ability to raise capital through alternative financing sources on terms acceptable to us, or at all, and may result in
the potential loss of confidence by investors, suppliers, customers and employees and fewer business development opportunities.
On
April 10, 2024, we received the Letter from the Staff of The Nasdaq Stock Market LLC indicating that, based upon the closing bid price
of our common stock for the 30 consecutive business days between February 27, 2024 and April 9, 2024, we did not meet the minimum bid
price of $1.00 per share required for continued listing on Nasdaq pursuant to the Bid Price Rule. The Letter also indicated that we were
provided with a compliance period of 180 calendar days, or until October 7, 2024, in which to regain compliance with the Bid Price Rule
pursuant to Nasdaq Listing Rule 5810(c)(3)(A). We did not regain compliance with the Bid Price Rule by October 7, 2024, and on October
8, 2024, Nasdaq notified us that our securities were subject to delisting from Nasdaq unless we timely requested a hearing before the
Panel. We subsequently and timely requested a hearing before the Panel, which was held on December 5, 2024.
On
November 19, 2024, we received an additional deficiency notice from the Staff indicating that we no longer satisfied the $2.5 million
stockholders’ equity requirement set forth in the Equity Rule for continued listing on Nasdaq. The Staff indicated that our non-compliance
with the Equity Rule would be considered by the Panel at the Hearing and could serve as an additional basis for delisting of our securities
from Nasdaq.
On
December 26, 2024, we received a letter (the “Decision Letter”) from the Panel granting a limited extension of time for us
to demonstrate compliance with the Bid Price Rule and the Equity Rule for continued listing on Nasdaq, subject to the following conditions:
(i) on or before February 27, 2025, we will have obtained stockholder approval to effect a reverse stock split of our common stock; (ii)
on or before March 31, 2025, we shall have effected a reverse stock split and, thereafter, maintain a $1.00 closing bid price of our
common stock for a minimum of ten consecutive trading days; (iii) on or before March 31, 2025, we are required to demonstrate compliance
with the Equity Rule by filing public disclosure with the SEC and demonstrate long-term compliance with the Equity Rule; and (iv) on
or before March 31, 2025, we are required to demonstrate compliance with all continued listing requirements for Nasdaq. On February 24,
2025, we obtained approval from our stockholders to file a certificate of amendment to our Certificate of Incorporation to effectuate
the March 2025 Reverse Stock Split, among others, and on March 13, 2025, the March 2025 Reverse Stock Split became effective.
On
April 9, 2025, we received a letter (the “April Letter”) from the Staff notifying us that we had demonstrated compliance
with the Bid Price Rule and the Equity Rule as required by the Panel pursuant to the Decision Letter.
Pursuant
to the April Letter, we will be subject to a mandatory panel monitor for a period of one year from the date of the April Letter. If,
within that one-year monitoring period, Staff finds us again out of compliance with the Equity Rule that was subject of the exception,
notwithstanding Nasdaq Listing Rule 5810(c)(2), we will not be permitted to provide the Staff with a plan of compliance with respect
to that deficiency and Staff will not be permitted to grant additional time for us to regain compliance with respect to that deficiency,
nor will the company be afforded an applicable cure or compliance period pursuant to Rule 5810(c)(3). Instead, the Staff will issue a
Delist Determination Letter and we will have an opportunity to request a new hearing with the Panel or a newly convened Hearings Panel
if the initial Panel is unavailable.
There
is no assurance that we will maintain compliance with such minimum listing requirements if we regain compliance with all applicable requirements
for continued listing on Nasdaq. If our common stock were delisted from Nasdaq, trading of our common stock would most likely take place
on an over-the-counter market established for unlisted securities, such as the OTCQB or the Pink Market maintained by OTC Markets Group
Inc. An investor would likely find it less convenient to sell, or to obtain accurate quotations in seeking to buy, our common stock on
an over-the-counter market, and many investors would likely not buy or sell our common stock due to difficulty in accessing over-the-counter
markets, policies preventing them from trading in securities not listed on a national exchange or other reasons. In addition, as a delisted
security, our common stock would be subject to SEC rules as a “penny stock,” which impose additional disclosure requirements
on broker-dealers. The regulations relating to penny stocks, coupled with the typically higher cost per trade to the investor of penny
stocks due to factors such as broker commissions generally representing a higher percentage of the price of a penny stock than of a higher-priced
stock, would further limit the ability of investors to trade in our common stock. In addition, delisting could harm our ability to raise
capital through alternative financing sources on terms acceptable to us, or at all, and may result in the potential loss of confidence
by investors, suppliers, customers and employees and fewer business development opportunities. For these reasons and others, delisting
would adversely affect the liquidity, trading volume and price of our common stock, causing the value of an investment in us to decrease
and having an adverse effect on our business, financial condition and results of operations, including our ability to attract and retain
qualified employees and to raise capital.
The
Certificate of Designations of the Series H Convertible Preferred Stock (“Series H Certificate of Designations”) provides
for the payment of cumulative dividends in shares of our common stock which will require us to have shares of common stock available
to pay the dividends.
Each
share of the Series H Convertible Preferred Stock (the “Series H Preferred Stock”) is entitled to receive cumulative dividends
at the rate per share of 9% per annum (as a percentage of the stated value per share), payable on each conversion date (with respect
to only the shares of Series H Preferred Stock being converted), in duly authorized, validly issued, fully paid and non-assessable shares
of common stock at the conversion price then on effect in accordance with the terms of the Certificate of Designations. As such, we may
rely on having available shares of common stock to pay such dividends, which will result in dilution to our shareholders. If we do not
have such available shares, we may not be able to satisfy our obligations as related to these dividends pursuant to the terms of the
Certificate of Designations.
The
Series G Certificate of Designations and the Series H Certificate of Designations both contain certain anti-dilution provisions, which
may dilute the interests of our stockholders, depress the price of our common stock, and make it difficult for us to raise additional
capital.
Certain
events, for example, a Dilutive Issuance (as defined in each of the Series G Certificate of Designations and the Series H Certificate
of Designations) may reduce the conversion price of the Series G Preferred Stock and/or Series H Preferred Stock, as applicable, which
in turn may lead to further dilution to the holders of our common stock. In addition, the perceived risk of dilution may cause our shareholders
to be more inclined to sell their common stock, which may in turn depress the price of common shares regardless of our business performance.
We may also find it more difficult to raise additional equity capital while any of the shares of the Series G Preferred Stock or the
Series H Preferred Stock remain outstanding.
Under
the Series H Purchase Agreement and the September 2025 Purchase Agreement, we are subject to certain restrictive covenants that may make
it difficult to procure additional financing.
Both
the Series H Purchase Agreement and the September 2025 Purchase Agreement provide that until the one year anniversary of the date of
the applicable closing contemplated by such agreement, we shall be prohibited from entering into any variable rate transactions, subject
to certain exceptions. If we require additional funding while this restrictive covenants remain in effect, we may be unable to effect
a financing transaction on terms acceptable to us, or at all, while also remaining in compliance with the terms of the Series H Purchase
Agreement and the September 2025 Purchase Agreement, or we may be forced to seek a waiver from the investor party to such agreements,
which such investor is not obligated to grant to us.
Mandatory
conversion of Series G Preferred Stock on Mandatory Conversion Date can result in substantial dilution to our existing shareholders.
Pursuant
to the terms of the Series G Certificate of Designations, on the fifth (5th) anniversary of the issuance date (the “Mandatory
Conversion Date”), all then outstanding shares of Series G Preferred Stock and, to the extent that we elect to pay dividends in
duly authorized, validly issued, fully paid and non-assessable shares of common stock, all accrued but unpaid dividends thereon through
and including the Mandatory Conversion Date shall be automatically converted into shares of common stock, subject to beneficial ownership
limitations applicable to each holder.
Such
mandatory conversion could result in substantial dilution to our existing shareholders, including a significant increase in the number
of shares of our common stock outstanding, which may reduce the ownership percentage and voting power of existing stockholders. In addition,
the issuance of a large number of shares of common stock upon conversion, or the expectation that such issuance may occur, could adversely
affect the market price of our common stock and increase volatility in our trading price.
Furthermore,
to the extent that at the time of such mandatory conversion we do not have a sufficient number of authorized but unissued and unreserved
shares of common stock to effect the conversion in full, we may be required to seek stockholder approval to increase our authorized share
capital, which may not be obtained in a timely manner or at all. Any delay in obtaining such approval could result in a failure to timely
satisfy our obligations under the Series G Preferred Stock, potentially constituting a breach of the applicable Certificate of Designations
and giving rise to additional rights or remedies for holders, including potential penalties or adjustments that could be adverse to us.
Even
if sufficient authorized shares are available, effecting such mandatory conversion will reduce the number of shares of common stock available
for future issuances, including for financings, acquisitions, equity incentive plans, or other corporate purposes. This reduction could
limit our flexibility to raise additional capital or pursue strategic transactions on favorable terms, or at all, which could adversely
affect our business, financial condition, and results of operations.
Future
issuances of preferred stock may adversely affect the market price for our common stock.
Additional
issuances and sales of preferred stock, or the perception that such issuances and sales could occur, may cause prevailing market prices
for our common stock to decline and may adversely affect our ability to raise additional capital in the financial markets at times and
prices favorable to us.
We
are a smaller reporting company, and we cannot be certain if the reduced disclosure requirements applicable to our filing status will
make our common stock less attractive to investors.
We
are a “smaller reporting company” and, thus, have certain decreased disclosure obligations in our SEC filings, including,
among other things, simplified executive compensation disclosures and only being required to provide two years of audited financial statements
in annual reports. Decreased disclosures in our SEC filings due to our status as a “smaller reporting company” may make it
harder for investors to analyze our results of operations and financial prospects and may make our common stock a less attractive investment.
If some investors find our common stock less attractive, there may be a less active trading market for our common stock and our stock
price may be more volatile.
Although
our shares of common stock are listed on Nasdaq, we currently have a limited trading volume, which results in higher price volatility
for, and reduced liquidity of, our common stock.
Although
our shares of common stock are listed on Nasdaq under the symbol “FEED,” trading volume in our common stock has been limited
and an active trading market for our shares of common stock may never develop or be maintained. The absence of an active trading market
increases price volatility and reduces the liquidity of our common stock. As long as this condition continues, the sale of a significant
number of shares of common stock at any particular time could be difficult to achieve at the market prices prevailing immediately before
such shares are offered.
Anti-takeover
provisions of our certificate of incorporation, our bylaws and Delaware law could make an acquisition of us, which may be beneficial
to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove the current members of our board
and management.
Certain
provisions of our amended and restated certificate of incorporation and bylaws could discourage, delay or prevent a Merger, acquisition
or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium
for your shares. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove members of
our Board of Directors (the “Board” or “Board of Directors”). These provisions also could limit the price that
investors might be willing to pay in the future for our securities, thereby depressing the market price of our securities. Stockholders
who wish to participate in these transactions may not have the opportunity to do so. These provisions, among other things:
| ● |
allow
the authorized number of directors to be changed only by resolution of our Board; |
| ● |
authorize
our Board to issue, without stockholder approval, preferred stock, the rights of which will be determined at the discretion of the
Board and that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer
to prevent an acquisition that our Board does not approve; |
| ● |
establish
advance notice requirements for stockholder nominations to our Board or for stockholder proposals that can be acted on at stockholder
meetings; and |
| ● |
limit
who may call a stockholder meeting. |
In
addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law that may, unless certain criteria
are met, prohibit large stockholders, in particular those owning 15% or more of the voting rights on our common stock, from merging or
combining with us for a prescribed period of time.
If
securities or industry analysts do not publish research or reports or publish unfavorable research about our business, the price of our
securities and their trading volume could decline.
The
trading market for our securities will depend in part on the research and reports that securities or industry analysts publish about
us or our business. Currently there is only one research coverage by a securities and industry analyst. If one or more of the analysts
who covers us downgrades our securities, the price of our securities would likely decline. If one or more of these analysts ceases to
cover us or fails to publish regular reports on us, interest in the purchase of our securities could decrease, which could cause the
price of our securities and their trading volume to decline.
Because
we do not expect to pay cash dividends for the foreseeable future, you must rely on appreciation of our common stock price for any return
on your investment. Even if we change that policy, we may be restricted from paying dividends on our common stock.
We
do not intend to pay cash dividends on shares of our common stock for the foreseeable future. Any determination to pay dividends in the
future will be at the discretion of our Board and will depend upon results of operations, financial performance, contractual restrictions,
restrictions imposed by applicable law and other factors our Board deems relevant. Accordingly, you will have to rely on capital appreciation,
if any, to earn a return on your investment in our common stock. Investors seeking cash dividends in the foreseeable future should not
purchase our common stock.
Our
ability to use our net operating loss carry forwards and certain other tax attributes may be limited.
Our
ability to utilize our federal net operating loss, carry forwards and federal tax credit may be limited under Sections 382 and 383 of
the Internal Revenue Code of 1986, as amended. The limitations apply if an “ownership change,” as defined by Section 382,
occurs. Generally, an ownership change occurs if the percentage of the value of the stock that is owned by one or more direct or indirect
“five percent shareholders” increases by more than 50% over their lowest ownership percentage at any time during the applicable
testing period (typically three years). If we have experienced an “ownership change” at any time since our formation, we
may already be subject to limitations on our ability to utilize our existing net operating losses and other tax attributes to offset
taxable income. In addition, future changes in our stock ownership, which may be outside of our control, may trigger an “ownership
change” and, consequently, Section 382 and 383 limitations. As a result, if we earn net taxable income, our ability to use our
pre-change net operating loss carry forwards and other tax attributes to offset U.S. federal taxable income may be subject to limitations,
which could potentially result in increased future tax liability to us.
If
we fail to maintain effective internal control over financial reporting, our business, financial condition or results of operations may
be adversely affected.
As
a public reporting company, we are required to establish and maintain effective internal control over financial reporting. Failure to
establish such internal control, or any failure of such internal control once established, could adversely impact our public disclosures
regarding our business, financial condition or results of operations. Any failure of our internal control over financial reporting could
also prevent us from maintaining accurate accounting records and discovering accounting errors and financial frauds.
Rules
adopted by the Securities and Exchange Commission pursuant to Section 404 of Sarbanes-Oxley Act of 2002 require annual assessment of
our internal control over financial reporting. The standards that must be met for management to assess the internal control over financial
reporting as effective are complex, and require significant documentation, testing and possible remediation to meet the detailed standards.
We may encounter problems or delays in completing activities necessary to make an assessment of our internal control over financial reporting.
If we cannot assess our internal control over financial reporting as effective, investor confidence and share value may be negatively
impacted. In addition, management’s assessment of internal control over financial reporting may identify weaknesses and conditions
that need to be addressed in our internal control over financial reporting or other matters that may raise concerns for investors. Any
actual or perceived weaknesses and conditions that need to be addressed in our internal control over financial reporting (including those
weaknesses identified in our periodic reports), or disclosure of management’s assessment of our internal control over financial
reporting may have an adverse impact on the price of our securities.
As
disclosed in Part II, Item 9A, “Controls and Procedures,” in this Annual Report on Form 10-K, we have identified material
weaknesses in our internal control over financial reporting due to lack of adequate controls over management’s review procedures
for processing, recording and reviewing transactions related to certain contracts, accounting memos and certain monthly closing procedures.
Therefore, we concluded that our internal control over financial reporting and related disclosure controls and procedures were not effective
as of December 31, 2025.
Risks
Related to the ENvue System
If
we are not successful in enhancing awareness of our ENvue System, driving adoption across our current target population and expanding
the population of eligible patients, our sales, business, financial condition and results of operations will be negatively affected.
Our
business currently depends primarily on our ability to successfully market our ENvue System, which involves successfully engaging with
group purchasing organizations or GPOs (i.e. entities that assist healthcare providers, such as hospitals, nursing homes, and home health
agencies, to achieve savings and efficiency by using aggregate purchasing volume to negotiate discounts with manufacturers, distributors,
and other suppliers) to increase adoption of and utilization our ENvue System.
The
medical community’s awareness of performing the feeding tube insertion procedure using our product and the medical community’s
adoption of the solution offered by us, instead of existing methods and products in the market for performing the procedure, is significant
and crucial for our success. We are aiming to increase awareness about our ENvue System, work with medical professionals in the United
States to raise awareness among the medical community and grow the number of facilities that utilize our ENvue System, but there can
be no assurance that we will succeed.
Our
commercial success and revenues will depend on the future adoption of the ENvue System into patient work streams in facilities and other
healthcare settings. If we are unable to successfully drive interest in our ENvue System, our business, financial condition and results
of operations would be harmed.
Our
commercial success and revenues will depend in large part on the future adoption of the ENvue System into patient work streams in facilities
and other healthcare settings. Our revenues are based and are expected to be based on the sale of the ENvue System and the sale of the
dedicated feeding tubes (which are consumable products) to hospitals. Hospital procurement budgets, including capital equipment budgets,
are sometimes shared by the entire institution or several departments within it. As such, expenses related to the purchase of other equipment
by certain departments of a medical institution may reduce the budgets available for the purchase of our products by other departments
interested in purchasing them.
The
commercial success of our ENvue System will continue to depend on a number of factors, including the following:
| ● |
the
actual and perceived effectiveness and clinical benefit, of our ENvue System; |
| |
|
| ● |
the
prevalence and severity of any adverse patient events involving our ENvue System; |
| |
|
| ● |
our
ability to provide earlier awareness of and education about our ENvue System to GPOs; |
| |
|
| ● |
the
degree to which medical professionals and GPOs adopt our ENvue System; |
| |
|
| ● |
the
availability, relative cost and perceived advantages and disadvantages of alternative technologies or treatment methods for cognitive
disorders; |
| |
|
| ● |
the
results of future clinical and other studies relating to the health, economic or other benefits of our ENvue System; |
| |
|
| ● |
whether
key thought leaders in the medical community accept that our future clinical utility is sufficiently meaningful to influence their
decision to adopt our ENvue System; |
| |
|
| ● |
the
extent to which we are successful in educating medical professionals, GPOs and patients about the benefits of our ENvue System; |
| |
|
| ● |
our
reputation among GPOs and medical professionals; |
| ● |
our
ability to predict product performance; |
| |
|
| ● |
the
strength of our marketing and distribution infrastructure, including our ability to drive adoption and utilization of our ENvue System; |
| |
|
| ● |
our
ability to obtain, maintain, protect, enforce and defend our intellectual property rights, including those covering our ENvue System; |
| |
|
| ● |
our
ability to maintain compliance with all legal and regulatory requirements, including FDA medical device postmarket surveillance regulations
applicable to our ENvue System; and |
| |
|
| ● |
our
ability to continue to attract and retain key talent. |
If
we fail to market and sell our ENvue System cost-effectively, our sales, business, financial condition and results of operations will
be negatively affected.
If
we are unable to successfully scale our marketing, training and quality control systems our business, financial condition and results
of operations would be harmed.
We
began our marketing and sales activities of ENvue System in the beginning of 2020, and as of the date of this filing, we have not yet
begun large-scale production and marketing. Accordingly, the use of the ENvue System has not yet been tested and proven on a large commercial
scale. We are in a continuous process of receiving feedback from product users, developing, and improving products, distributing the
improved products, and continuously reviewing our training procedures and quality control. We estimate that we will need to develop our
marketing, training, and quality control systems to a scale not currently available to us (or alternatively, enter into an agreement
with a strategic distributor or marketer who has such capabilities). There is no assurance that we will be able to do so in a way that
allows us to achieve our objectives. If we are unable to successfully implement such marketing, training and quality control systems
our business, financial condition and results of operations would be harmed.
We
may be unable to compete successfully with competitive technologies, which could harm our sales, business, financial condition and results
of operations.
Our
industry is competitive and has been evolving rapidly. As of February 13, 2019, the ENvue System and ENvue Feeding Tube has received
FDA clearance for marketing in the U.S. under the 510(k) procedure for use in adults (age 22 and older). As we continue to engage with
target GPOs to increase adoption and utilization of our ENvue System, we expect to face competition in the market from competing technologies,
as well as competition from new companies that may enter the market or introduce new technologies in the future. Third-party payors may
encourage the use of competitors’ products due to lower costs of competing products or alternatives. Additionally, treating physicians
may promote the use of other competitors’ products or alternative therapies.
Our
current and future competitors may include large, well-capitalized companies with significant market share and resources. They may have
more established sales and marketing programs than we do and have greater name recognition. In addition to competing for market share,
competitors may develop or acquire patents or other rights that may limit our ability to compete.
We
believe that the competitive advantages of our ENvue System will be important factors in our future success. Our continued success depends
on, among other things, our ability to:
| ● |
successfully
engage with GPOs to increase adoption of and utilization our ENvue System; |
| |
|
| ● |
attract
and retain skilled research, development, sales, marketing and clinical personnel; |
| |
|
| ● |
continue
to innovate in order to improve our ENvue System and enhance the patient and provider experience; |
| |
|
| ● |
adequately
predict and respond to product performance and safety; |
| |
|
| ● |
obtain
and maintain regulatory clearances, including for expanded indications; |
| |
|
| ● |
cost-effectively
market and sell our ENvue System; |
| |
|
| ● |
obtain,
maintain, protect, enforce and defend our intellectual property rights and operate our business without infringing, misappropriating
or otherwise violating the intellectual property rights of others; and |
| |
|
| ● |
acquire
products or technologies complementary to or necessary for our business. |
The
medical device industry is intensely competitive, subject to rapid change and significantly affected by new product introductions and
other market activities of industry participants. There can be no assurance that other companies or institutions will not succeed in
developing or marketing devices and products that are more effective than our ENvue System or that would render our ENvue System obsolete
or noncompetitive.
The
misuse or off-label use of our ENvue System may harm our reputation in the marketplace, result in injuries that lead to product liability
suits or result in costly investigations, fines or sanctions by regulatory bodies, particularly if we are deemed to have engaged in the
promotion of these uses, any of which could be costly to our business.
Our
ENvue System is a Class II medical device cleared by FDA for commercialization in the U.S. to aid qualified operators in the placement
of ENvue Medical Enteral Feeding Tube into the stomach or small intestine pursuant to the 510(k) notification process in February 2019
for use in adults (aged 22 and over). We, thus, are not currently able to promote the ENvue System for any other indications for use
or make any promotional claims that are inconsistent with, or outside the scope of, such FDA clearance (often referred to as “off-label”
claims). However, the assessment of whether a given claim is or is not consistent with a given FDA clearance or approval can often be
subjective, and we cannot guarantee that FDA will always agree with our position regarding a particular claim or that all of our employees,
representatives, and agents will abide by our marketing policies. If FDA determines that we have promoted any product without the requisite
clearance or approval and/or for an off-label or unapproved use, it could take any number of enforcement actions against us, including
(among others), issuing untitled or warning letters and/or pursuing an injunction, seizure, civil fine and/or criminal penalties. It
is also possible that other federal, state or foreign enforcement authorities might take action under other regulatory authority, such
as laws prohibiting false claims for reimbursement, any of which would have a material adverse effect on our financial condition and/or
business as a whole.
Additionally,
we must have competent and reliable scientific evidence or, where applicable, other adequate substantiation for each reasonable interpretation
of every promotional claim we make. In particular, comparative or superiority claims generally require adequate, well controlled, head-to-head
clinical studies, comparing the product to the applicable competing products. To the extent we make any claims, or are otherwise held
responsible for third-party claims about any product we may market in the United States, without the requisite clinical substantiation,
we could be subject to enforcement action by FDA and/or the Federal Trade Commission (FTC), as well as a competitor challenge via the
National Advertising Division (NAD) of the Better Business Bureau. Our plans to utilize social media as a primary promotional tool for
our device(s) increases the applicable enforcement risk, as it makes it easier for our employees, affiliates, and any third parties with
which we may have a relationship and/or arrangement under which we are deemed responsible for such party’s claims about our product(s)
to disseminate promotional claims about our product(s) that may be inconsistent with applicable regulations governing device promotions.
Further, consumers can bring private false-advertising lawsuits, including class actions, against us for any material misrepresentations
and/or deceptive or unsubstantiated claims (among other similar causes of action) in our promotional materials or other advertising.
Any of the foregoing could have a material adverse effect on our business.
Use
of our ENvue System requires appropriate training and inadequate training may lead to negative clinician experiences, which could harm
our business, financial condition, and results of operations.
The
successful use of our ENvue System depends in part on the training and skill of the clinician. According to the regulatory clearance
of the ENvue System by the FDA, users of the system are required to undergo training provided by us, according to a unique and easy-to-implement
training model developed by us for system users. The training is usually provided in a concentrated manner to system users on behalf
of the hospital, lasts approximately five days, and includes both theoretical and practical components regarding the system and its use.
Providers could experience difficulty using our ENvue System. Moreover, medical providers rely on their previous medical training and
experience when recommending or utilizing our ENvue System, and we cannot guarantee that all clinicians will have the necessary skills
to properly utilize the ENvue System. We cannot be certain that clinicians that will use our ENvue System will have received sufficient
training, and clinicians who have not received adequate training may nonetheless attempt to use our ENvue System with their patients.
If medical providers utilize our ENvue System incorrectly, or without adhering to or completing all relevant training, their patient
outcomes may not be consistent with the outcomes achieved in our research studies and any future clinical studies. Adverse safety outcomes
that arise from improper or incorrect use of our ENvue System may negatively impact the perception of patient benefit and the safety
of our ENvue System, notwithstanding results from our research studies and any future clinical studies. These results could limit adoption
of our ENvue System, which would harm our sales, business, financial condition, and results of operations.
Our
business could be adversely affected by professional and legal challenges to our business model or by new state actions restricting our
ability to provide our products and services in certain states.
Since
the success of our business will be dependent on the widespread adaptation of our ENvue System as an efficient and safer solution for
feeding tube insertion compared to the alternative methods currently available on the market, clinicians and medical professionals across
multiple geographies will be needed to use our ENvue System and provide positive feedback and results. This will expose the Company to
legal risk of patients or medical providers who may have a negative experience with our ENvue System filing lawsuits claiming damages
or other claims. Although the Company will seek insurance coverage for such legal actions, there is no assurance that the amount of coverage
will be sufficient to cover these claims. In addition, such legal actions from consumers and medical providers may result in material
and adverse effects on our ability to continue to conduct business due to negative press.
We
may not be able to achieve or maintain satisfactory pricing and margins for our ENvue System, which could harm our business and results
of operations.
The
medical device industry has a history of price competition, and we can give no assurance that we will be able to maintain satisfactory
prices for our ENvue System or any future products at competitive levels. The pricing of our products could be impacted by several factors,
including change of supplies, price changes of components, and shipping costs. If we are forced to lower or are unable to increase the
price we charge for our ENvue System, our gross margins will decrease, which will harm our ability to invest in and grow our business.
If we are unable to maintain our prices, or if our costs increase and we are unable to offset such increase with an increase in our prices,
our margins could erode, which could harm our business and results of operations.
Future
sales of our ENvue System may depend on providers’ and patients’ ability to obtain reimbursement from third-party
payors, such as insurance carriers.
Future
sales of our ENvue System may depend on our provider customers’ and patients’ ability to obtain reimbursement from third-party
payors, such as insurance carriers. Our customers typically rely significantly on insurance or third-party reimbursement for the treatment
and care they provide to patients. Any reduction in insurance or other third-party payor reimbursement for such patient care may cause
negative price pressure that affects their ability to purchase our ENvue System, which would reduce our revenues. Without a corresponding
reduction in the cost to produce such products, the result would be a reduction in our overall gross profit. Similarly, any increase
in the cost of such products would likely reduce our overall gross profit unless there was a corresponding increase in third-party payor
reimbursement. Failure by our provider customers or their patients to obtain or maintain coverage or to secure adequate reimbursement
for treatment by third-party payors could have an adverse effect on our business, results of operations, and financial condition.
Our
results of operations may be harmed if we are unable to accurately forecast clinician demand for our ENvue System or any future products.
Our
ability to accurately forecast demand for our ENvue System or our future our products could be negatively affected by many factors, including
our failure to accurately manage our expansion strategy, product introductions by competitors, our inability to forecast the lifecycle
of our products, an increase or decrease in customer demand for our products or for competitor products, our failure to accurately forecast
customer adoption of new products, unanticipated changes in general market conditions or regulatory matters and weakening of economic
conditions or consumer confidence in future economic conditions. For us to succeed, it is essential to introduce and integrate the ENvue
System into our target market, including through the creation of strategic partnerships and the establishment of effective marketing
and distribution networks, as well as the successful execution of commercial validation of the ENvue System. Inventory levels in excess
of customer demand may result in inventory write-downs or write-offs, which would cause our gross margin to be adversely affected and
could impair the strength of our brand, which may negatively affect our business, financial condition, and results of operations.
Adoption
of our ENvue System depends on positive clinical data as well as medical providers’ acceptance of the data and our products, and
negative clinical data, publicly reported adverse events, or perceptions among these medical providers would harm our sales, business,
financial condition, and results of operations.
The
rate of adoption and sales of our products is heavily influenced by clinical data. There can be no assurance that future clinical studies,
including those to demonstrate the efficacy of our ENvue System or future products in current target patient populations and those to
support label retention and expansion for our products, will demonstrate clinical utility and effectiveness. Unfavorable or inconsistent
clinical data from future clinical studies conducted by us, our competitors, or third parties, adverse events publicly reported by us,
patients, or healthcare providers, or the negative interpretation of our clinical data internally and externally, including by customers,
competitors, patients, and regulators could harm our business, financial condition, and results of operations.
The
rate of adoption and sales of our products are also influenced by clinician perceptions. Negative perceptions of our products by medical
providers, including due to negative clinical data or adverse events, could result in decreased adoption or use of our products, which
would harm our business, financial condition, and results of operations. Further, if we are not able to attain strong working relationships
with medical providers and receive their advice and input, the marketing of our products could suffer, which could harm our business,
financial condition and results of operations.
Our
future success also depends upon patients having an experience with our products that meets their expectations in order to increase clinician
demand for our products as a result of positive feedback and word-of-mouth. Patients may experience negative clinical outcomes if the
performing medical providers are not adequately trained on use of our ENvue System. If the results of our products do not meet the expectations
of the patients or their providers, it could discourage continuing use of our device or referring our products to others. Dissatisfied
providers or patients may express negative opinions through social media, advocacy, or other publicity. Any failure to meet provider
or patient expectations and any resulting negative publicity could harm our reputation and future sales.
Risks
Related to the 2025 Reverse Stock Split
The
2025 Reverse Stock Split may not increase the price of our common stock over the long-term and our common stock may be delisted.
The
principal purpose of the 2025 Reverse Stock Split was to increase the trading price of our common stock to meet the minimum stock price
standards of Nasdaq. However, the effect of a reverse stock split on the market price of our common stock cannot be predicted with any
certainty, and we cannot assure you that a reverse stock split will accomplish this objective for any meaningful period of time, or at
all. While we expect that the reduction in the number of outstanding shares of common stock will proportionally increase the market price
of our common stock, we cannot assure you that a reverse stock split will increase the market price of our common stock by a multiple
of any reverse stock split ratio, or result in any permanent or sustained increase in the market price of our common stock sufficient
to regain compliance with the conditions required by the Panel. The market price of our common stock may be affected by other factors
which may be unrelated to the number of shares outstanding, including our business and financial performance, general market conditions,
and prospects for future success.
The
2025 Reverse Stock Split may decrease the liquidity of our common stock.
The
2025 Reverse Stock Split reduced the total number of outstanding shares of common stock, which may lead to reduced trading and a smaller
number of market makers for our common stock, particularly if the price per share of our common stock does not increase as a result of
a reverse stock split.
The
2025 Reverse Stock Split may result in some stockholders owning “odd lots” that may be more difficult to sell or require
greater transaction costs per share to sell.
The
2025 Reverse Stock Split had the effect of increasing the number of stockholders who own “odd lots” of less than 100 shares
of common stock. A purchase or sale of less than 100 shares of common stock (an “odd lot” transaction) may result in incrementally
higher trading costs through certain brokers, particularly “full service” brokers. Therefore, those stockholders who own
fewer than 100 shares of common stock following a reverse stock split may be required to pay higher transaction costs if they sell their
common stock.
ITEM
1B. UNRESOLVED STAFF COMMENTS
None.
ITEM
1C. CYBERSECURITY
Cybersecurity
Risk Management and Governance
We
operate in the medical device and healthcare technology sector, which is subject to various cybersecurity risks that could adversely
affect our business, financial condition, and results of operations, including intellectual property theft; fraud; extortion; harm to
employees or customers; violation of privacy laws and other litigation and legal risk; and reputational risk. We recognize the critical
importance of developing, implementing, and maintaining cybersecurity measures to safeguard our information systems and protect the confidentiality,
integrity, and availability of our data.
Cybersecurity
Risk Assessment Program
Our
cybersecurity risk assessment program consists of an annual review of our risks and policies. The program outlines governance, policies
and procedures, and technology controls we use to oversee and identify risks from cybersecurity threats, and is informed by cybersecurity
incidents we have observed both within the Company and in our industry. We are adopting formal policies governing information security,
acceptable use, and data retention, which are reviewed and updated on at least an annual basis.
We
have engaged a third party virtual Chief Information Security Officer (CISO) to assist in assessing, developing, and maintaining our
cybersecurity program. Among other things, this engagement includes periodic evaluations of our workstations and on site storage equipment.
We
maintain and constantly improve security measures designed to protect client, patient, customer, employee, and vendor information and
to prevent data loss and other security breaches. We maintain business continuity, contingency, and recovery plans for use in the event
of a cybersecurity incident, including local and cloud based backup of files and emails.
Third
Party Service Provider Risk
We
have implemented processes to oversee and identify cybersecurity risks associated with our use of third party service providers. These
processes include vendor due diligence prior to engagement, contractual requirements for security standards where appropriate, and ongoing
monitoring of compliance with applicable protocols. We require that third party software providers used for accounting, billing, and
payroll maintain SOC 1 compliance, and we monitor their continued adherence to those standards.
Cross-Border
Operations
We
maintain operations in both the United States (Tyler, Texas, Arlington Heights, Illinois) and Israel (Tel Aviv), including through our
subsidiary NanoVibronix Ltd. (Nesher, Tel-Aviv) Our cybersecurity program is designed to address the risks associated with cross-border
data flows and the applicable data protection requirements in each jurisdiction, including Israeli privacy law. We seek to ensure that
our policies and controls are applied consistently across both locations.
Management
and Board Oversight
Our
Chief Operating Officer, with oversight from the Chief Executive Officer and senior management, is responsible for the day-to-day assessment
and management of risks from cybersecurity threats, including the prevention, mitigation, detection, and remediation of cybersecurity
incidents. Day-to-day operational cybersecurity activities are managed by our IT Manager in coordination with external IT support.
The
Audit Committee of the Board of Directors is responsible for oversight of risks from cybersecurity threats in conjunction with management.
The Committee receives interim reports and updates from senior management, and management has committed to updating the full Board on
a quarterly basis with respect to the management of risks from cybersecurity threats. Such reports cover our information technology security
program, including its current status, capabilities, objectives, and plans, as well as the evolving cybersecurity threat landscape. The
Nominating and Corporate Governance Committee considers risks from cybersecurity threats as part of its oversight of the Company’s
business strategy, risk management, and financial oversight.
Materiality
Assessment
In
determining whether a cybersecurity incident is material, senior management evaluates the nature and scope of the incident, the data
or systems affected, potential financial impact, regulatory implications, and any operational disruption. Material incidents are escalated
to the Chief Executive Officer and reported to the Nominating and Corporate Governance Committee in accordance with our incident response
procedures.
As
of the date of this report, no cybersecurity incident (or aggregation of incidents) or cybersecurity threat has materially affected our
results of operations or financial condition. However, an actual or perceived breach of our security could damage our reputation, cause
existing clients and customers to discontinue their relationships with us, prevent us from attracting new clients and customers, interfere
with the progress of our clinical trials, or interfere with our efforts to pursue regulatory approvals for our product candidates, or
subject us to third party lawsuits, regulatory fines, or other actions or liabilities, any of which could adversely affect our business,
operating results, or financial condition. For further information, see “Risk Factors-Our business and operations would suffer
in the event of computer system failures, cyber-attacks or deficiencies in our cyber-security” in Item 1A of this Annual Report
on Form 10-K. We currently carry a cyber liability insurance policy.
ITEM
2. PROPERTIES
We
lease office and manufacturing facilities in Nesher, Israel, maintain a corporate office in Tel Aviv, Israel through ENvue, and maintain an office in Tyler, Texas.
In
March 2024, we entered into a three-year lease for an office and manufacturing facility in Nesher, Israel. The lease provides for monthly
rent of approximately $2,500 and covers approximately 180 square meters. The lease includes an option to terminate the agreement at any
time after the first twelve months upon four months’ prior written notice.
We also maintain an office in Tyler, Texas, for which
we pay approximately $1,200 per month. This arrangement is currently on a month-to-month basis and is not subject to a formal long-term
lease agreement.
We also have ENvue’s offices and facilities in Tel Aviv,
Israel, consisting of approximately 490 square meters, with monthly rent of approximately $4,900.
In addition,
ENvue has an agreement for offices and facilities located in Arlington Heights, Illinois, for storage, inventory management,
order processing, and shipping services within the United States, for which we pay approximately $4,000 per month.
We
believe our existing facilities and logistics arrangements are adequate to meet our current operational needs and anticipated near-term
growth.
ITEM
3. LEGAL PROCEEDINGS
From
time to time, we may be involved in certain claims and litigation arising out of the ordinary course and conduct of business. Management
assesses such claims and, if it considers that it is probable that an asset had been impaired or a liability had been incurred and the
amount of loss can be reasonably estimated, provisions for loss are made based on management’s assessment of the most likely outcome.
Protrade
Proceeding
On
February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the
International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach
of an Exclusive Distribution Agreement dated March 7, 2019 (the “Agreement”) between Protrade and the Company. Protrade alleges,
in part, that the Company has breached the Agreement by discontinuing the manufacture of the DV0057 Painshield MD device in favor of
an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million. The Company vigorously defended the claims
asserted by Protrade.
On
March 15, 2022, the arbitrator issued a final award, which, determined that (i) the Company had the right to terminate the Exclusive
Distribution Agreement; (ii) the Company did not breach the duty of good faith and fair dealing with regard to the Exclusive Distribution
Agreement; and (iii) the Company did not breach any confidentiality obligations to Protrade. Nevertheless, the arbitrator determined
that the Company did not comply with the obligation to supply Protrade with a year’s supply of patches, and awarded Protrade $1,500,250,
which consists of $1,432,000 for “lost profits” and $68,250 as reimbursement of arbitration costs, on the grounds that the
Company allegedly failed to supply Protrade with certain patches utilized by users of DV0057 Painshield MD device. The arbitrator based
the decision on the testimony of Protrade’s president who asserted that a user would use in excess of 33 patches per each device.
The Company believes that the number of patches per device alleged by Protrade is grossly inflated, and that these claims were not properly
raised before the arbitrator. Accordingly, on April 13, 2022, the Company submitted an application for the correction of the award which
the arbitrator denied on June 22, 2022.
On
April 5, 2022, Protrade filed a Petition with the Supreme Court of New York Nassau County seeking to confirm the Award. On April 13,
2022, the Company submitted an application to the ICA seeking to correct an error in the award based on the evidence that the Company
only sold 2-3 reusable patches per device contrary to the 33 reusable patches claimed by Protrade. The same arbitrator who issued the
award, denied the application.
On
July 22, 2022, the Company filed a cross-motion seeking to vacate arbitration award on the grounds that the arbitrator exceeded her authority,
that the award was procured by fraud, and that the arbitrator failed to follow procedures established by New York law. In particular,
the Company averred in its motion that Protrade’s witness made false statements in arbitration, and that the arbitrator resolved
a claim that was never raised by Protrade and that has no factual basis.
On
October 3, 2022, the court issued a decision granting Protrade its petition to confirm the award and denying the cross-motion.
On
November 9, 2022, the Company filed a motion to re-argue and renew its cross-motion to vacate the arbitration decision based on newer
information that was not available during the initial hearing. On the same day, the Company also filed a notice of appeal with the Appellate
Division, Second Department. On March 21, 2023, the court denied the motion to re-argue and renew.
On
July 10, 2023, the Company filed its appeal with the Appellate Division, Second Department. That appeal is now fully briefed. In February
2025, the Second Department informed counsel for the Company that the Second Department was beginning to process the appeal for calendaring.
On
March 30, 2026, the Appellate Division of the Second Department issued a decision and order which affirmed the judgment of the Supreme
Court, Nassau County in its entirety and dismissed the appeal, stating inter alia that “NanoVibronix failed to establish that the
arbitration award violated a strong public policy, was irrational, procured by fraud, or clearly exceeded a specifically enumerated limitation
of the arbitrator’s power”. It further held that NanoVibronix had not demonstrated that it had shown sufficient new facts
to the court on the motion to renew. The decision was conclusory and did not analyze the facts as argued by NanoVibronix nor did it distinguish
them specifically. The Company is currently reviewing the decision and considering its available alternatives.
As
of December 31, 2025, and 2024, the Company accrued the amount of the arbitration award to Protrade of approximately $2.3 and $2.1 million,
respectively, including interest which is classified in “Other accounts payable and accrued expenses”.
See
also “Item 8. Financial Statements and Supplementary Data - Note 12. Commitments and Contingencies,” which information is
incorporated herein by reference, for a description of pending and recent litigation.
ITEM
4. MINE SAFETY DISCLOSURES
Not
applicable.
PART
II
ITEM
5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market
Information
Our
common stock is listed on Nasdaq under the symbol “FEED.” Prior to December 12, 2025, our common stock was listed on Nasdaq
under the symbol “NAOV.” Prior to listing on Nasdaq, our common stock was quoted on the OTCQB over-the-counter marketplace
under the symbol “NAOV” since April 10, 2015. Prior to April 10, 2015, there was no established public trading market for
our common stock.
Related
Stockholder Matters
As
of March 31, 2026, we had 1,100,413 issued and outstanding shares of common stock, 820 issued and outstanding shares of Series G Preferred
Stock, 11,111 issued and outstanding shares of Series H Preferred Stock and 53,100 issued and outstanding shares of Series X Non-Voting
Convertible Preferred Stock. The common stock was held by 103 holders of record, the Series G Preferred Stock was held by
1 holder of record, the Series H Preferred Stock was held by 1 holder of record and the Series X Preferred
Stock was held by 6 holders of record. The actual number of holders of our common stock is greater than the number of record
holders, and includes stockholders who are beneficial owners, but whose shares are held in street names by brokers or other nominees.
Authorized
Capital and Preferred Stock
On
March 3, 2021, we filed a proxy statement in connection with a special meeting of stockholders that was held on March 31, 2021, and ultimately
adjourned until May 6, 2021, to (i) ratify the increase in the number of authorized shares of common stock from 20,000,000 to 24,109,635
and the issuance of such 4,109,635 shares of common stock, and (ii) further increase the number of our authorized shares of common stock.
On May 6, 2021, the Company’s stockholders voted to approve the ratification of the increase in the number of authorized shares
of common stock from 20,000,000 to 24,109,635 and the issuance of such 373,603 shares of common stock to be effective as of December
4, 2020, but the stockholders did not approve a further increase in the number of its authorized shares of common stock.
On
August 17, 2021, the Company’s stockholders voted to approve an amendment to our Amended and Restated Certificate of Incorporation
to increase the number of shares of our common stock authorized for issuance from 24,109,635 shares to 40,000,000 shares.
As
of March 31, 2026, there were no shares of our Series C Preferred Stock issued and outstanding. Each share of our Series C Preferred
Stock is convertible into one share of our common stock (subject to adjustment as provided in the related designation of preferences)
at any time at the option of the holder, provided that the holder would be prohibited from converting Series C Preferred Stock into shares
of our common stock if, as a result of such conversion, the holder, together with its affiliates, would beneficially own more than 9.99%
of the total number of shares of our common stock then issued and outstanding. This limitation may be waived upon not less than 61 days’
prior written notice to us.
As
of March 31, 2026, there were no shares of our Series D Preferred Stock outstanding. Each share of our Series D Preferred Stock is convertible
into one thousand shares of our common stock (subject to adjustment as provided in the related designation of preferences) at any time
at the option of the holder, provided that the holder would be prohibited from converting Series D Preferred Stock into shares of our
common stock if, as a result of such conversion, the holder, together with its affiliates, would own more than 9.99% of the total number
of shares of our common stock then issued and outstanding. This limitation may be waived upon not less than 61 days’ prior written
notice to us.
As
of March 31, 2026, there were no shares of our Series E Preferred Stock issued and outstanding. Each share of our Series E Preferred
Stock is convertible into one share of our common stock (subject to adjustment as provided in the related designation of preferences)
at any time at the option of the holder, provided that the holder would be prohibited from converting Series E Preferred Stock into shares
of our common stock if, as a result of such conversion, the holder, together with its affiliates, would beneficially own more than 9.99%
of the total number of shares of our common stock then issued and outstanding. This limitation may be waived upon not less than 61 days’
prior written notice to us.
As
of March 31, 2026, there were no shares of our Series F Preferred Stock issued and outstanding. Each share of Series F Preferred Stock
entitles the holder thereof to 1,000,000 votes per share (and, for the avoidance of doubt, each fraction of a share of Series F Preferred
Stock has a ratable number of votes). Thus, each one-thousandth of a share of Series F Preferred Stock entitles the holder thereof to
1,000 votes. The outstanding shares of Series F Preferred Stock will vote together with the outstanding shares of common stock of the
Company as a single class exclusively with respect to (1) any proposal to adopt an amendment to Certificate of Incorporation to reclassify
the outstanding shares of common stock at a ratio specified in or determined in accordance with the terms of such amendment and (2) any
proposal to adjourn any meeting of stockholders called for the purpose of voting on the matters mentioned in the aforementioned proposal.
The Series F Preferred Stock is not entitled to vote on any other matter, except to the extent required under the Delaware General Corporation
Law.
As
of March 31, 2026, there were 62,220 shares of Series X Preferred Stock authorized and 53,100 shares of our Series X Preferred
Stock issued and outstanding. As of March 31, 2026, the conversion price for each share of Series X Preferred Stock is $20.40. The
conversion ratio (the “Conversion Ratio”) for each share of Series X Preferred Stock is determined by dividing the
Stated Value (as defined in the Series X Certificate of Designations, as defined below) of each share of Series X Preferred Stock,
initially valued at $606.3756, divided by the conversion price which provides an implied Conversion Ratio of 1,000 shares of common
stock issuable upon the conversion of each share of Series X Preferred Stock, subject to adjustment as provided in the Certificate
of Designations of the Series X Non-Voting Convertible Preferred Stock (the “Series X Certificate of Designations”).
Effective as of 5:00 p.m. Eastern Time on the fourth business day after the approval of the shares of common stock issuable upon
conversion of the Series X Preferred Stock (the “Series X Stockholder Approval”), each share of Series X Preferred Stock
then outstanding shall automatically convert into a number of shares of common stock equal to the Conversion Ratio, subject to
applicable beneficial ownership limitations. Subject the terms of the Series X Certificate of Designations, the Series X Preferred
Stock is also convertible, at the option of the holder, at any time and from time to time following 5:00 p.m. Eastern Time on the
third business day after the date that the Series X Stockholder Approval, into a number of shares of common stock equal to the
Conversion Ratio, subject to the applicable beneficial ownership limitations. Except as otherwise provided in the Series X
Certificate of Designations, or as required by the DGCL, the Series X Preferred Stock shall have no voting rights. As of December
31, 2025, there were 820 shares of our Series G Preferred Stock issued and outstanding. The Series G Preferred Stock is convertible
into shares of common stock at a current conversion price equal to $17.77 per share of common stock, subject to adjustment as
provided in the Series G Certificate of Designations, at any time at the option of the holder prior to the fifth anniversary of the
date of issuance, at which time all shares of outstanding Series G Preferred Stock shall automatically and without any further
action by the holder be converted into shares of common stock at the then effective conversion price. The holders of Series G
Preferred Stock are entitled to receive cumulative dividends at the rate per share of 9% per annum of the stated value per share,
until the fifth anniversary of the date of issuance of the Series G Preferred Stock. The five year 9% per annum dividend will be
paid upon conversion of the Series G Preferred Stock irrespective of the timing of conversion such that upon conversion, the
conversion price will incorporate the five-year 9% dividend. Except as otherwise provided in the Series G Certificate of
Designations or as otherwise required by law, the Series G Preferred Stock shall have no voting rights.
As
of December 31, 2025, there were 11,111 shares of our Series H Preferred Stock issued and outstanding. The Series H Preferred Stock
were initially convertible into the common stock at the election of the holder at any time at an initial conversion price of $10.10 per
share, and following the additonal investment right consummated in March 24, 2026 and as March 31, 2026, at the current conversion price
of $1.0422 per share (the “Series H Conversion Price”). The Series H Conversion Price is subject to customary adjustments for
stock dividends, stock splits, reclassifications, stock combinations and the like (subject to certain exceptions) and subject to price-based
adjustment in the event of any issuances of common stock, or securities convertible, exercisable or exchangeable for common stock, at
a price below the then-applicable Series H Conversion Price (subject to certain exceptions). Except as otherwise provided in the Series
H Certificate of Designations or as otherwise required by law, the Series H Preferred Stock shall have no voting rights.
Recent
Sales of Unregistered Securities
All
sales of unregistered securities during the year ended December 31, 2025, were previously disclosed in a Quarterly Report on Form 10-Q
or a Current Report on Form 8-K.
Issuer
Purchases of Equity Securities
We
did not purchase any of our registered equity securities during the period covered by this Annual Report.
Dividends
We
have not paid any cash dividends to our stockholders since inception and do not plan to pay cash dividends in the foreseeable future.
Any future declaration of dividends will depend on our earnings, capital requirements, financial condition, prospects and any other factors
that our board of directors deems relevant, as well as compliance with the requirements of state law. In general, as a Delaware corporation,
we may pay dividends out of surplus capital or, if there is no surplus capital, out of net profits for the fiscal year in which a dividend
is declared and/or the preceding fiscal year. We currently intend to retain earnings, if any, for reinvestment in our business.
ITEM
6. RESERVED
ITEM
7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Management’s
Discussion and Analysis of Financial Condition and Results of Operations is intended to provide a reader of our financial statements
with a narrative from the perspective of our management on our financial condition, results of operations, liquidity, and certain other
factors that may affect our future results. You should read the following discussion and analysis of financial condition and results
of operations in conjunction with our consolidated financial statements and the related notes thereto included elsewhere in this Annual
Report on Form 10-K. In addition to historical information, the following discussion and analysis includes forward-looking information
that involves risks, uncertainties and assumptions. Our actual results and the timing of events could differ materially from those anticipated
by these forward-looking statements as a result of many factors, including those discussed under “Item 1A. Risk Factors”
and elsewhere in this Annual Report on Form 10 -K. See “Item 1. Business - Cautionary Note Regarding Forward-Looking Statement;
Risk Factors Summary” included elsewhere in this Annual Report on Form 10 -K.
Overview
We
were organized as a Delaware corporation in October 2003. On February 14, 2025, we completed the Merger pursuant to the Merger Agreement,
as further described below. Following the consummation of the Merger, NanoVibronix will conduct its operations through its two wholly-owned
subsidiaries: (i) NanoVibronix Ltd., a private company incorporated under the laws of the State of Israel (“Nano OpCo”) and
(ii) ENvue Medical Holdings LLC, a Delaware limited liability company (together with its respective subsidiaries, “ENvue”).
Nano OpCo focuses on non-invasive biological response-activating devices that target biofilm prevention, pain therapy, and wound healing
and can be administered at home, without the assistance of medical professionals. ENvue is a medical device company engaged in the research,
development, production, marketing, and sale of medical devices in the field of enteral feeding and are in the initial stage of commercializing
our products.
Agreement
and Plan of Merger
On
February 14, 2025, pursuant to the terms of that certain Agreement and Plan of Merger (the “Merger Agreement”), dated as
of February 14, 2025, by and among us, NVEH Merger Sub I, Inc., a Delaware corporation and a wholly-owned subsidiary of the Company (“First
Merger Sub”), NVEH Merger Sub II, LLC, a Delaware limited liability company and a wholly-owned subsidiary of the Company (“Second
Merger Sub”), and ENvue Medical Holdings, Corp. (“Predecessor ENvue” or “ENvue”), the Company and Predecessor
ENvue effected (i) a merger of First Merger Sub with and into Predecessor ENvue, with the First Merger Sub ceasing to exist and Predecessor
ENvue becoming a wholly-owned subsidiary the Company and (ii) the merger of Predecessor ENvue with and into Second Merger Sub (the “Second
Merger” and, together with the First Merger, the “ENvue Merger”), with Second Merger Sub being the surviving entity
of the Second Merger (“Surviving Entity”). At the effective time of the Second Merger, the certificate of formation of the
Surviving Entity was amended and restated to, among other things, to change the name of the Surviving Entity to “ENvue Medical
Holdings LLC.” In connection with the Merger Agreement, we issued (i) 3,318 shares of common stock (the “Merger Shares”),
which such number of shares represented no more than 4.9% of the outstanding shares of common stock as of immediately before the First
Effective Time and (ii) Pre-Funded Warrants to purchase up to 12,526 shares of our common stock (the “Merger Pre-Funded Warrants”)
at an exercise price of $0.001 per share, and (iii) 57,720 shares of Series X Non-Voting Convertible Preferred Stock (the “Series
X Preferred Stock”). In addition, the Company issued 3,626 shares of Series X Preferred
Stock to a service provider of ENvue, replacing its equity interest in ENvue, resulting in a total of 61,346 shares of Series
X Preferred Stock outstanding after the Merger.
Nasdaq
Minimum Stockholder’s Bid Price Requirement and Minimum Stockholder’s Equity Requirement
As
previously disclosed, on April 10, 2024, we received a letter (the “Letter”) from the Listing Qualifications Department (the
“Staff”) of The Nasdaq Stock Market LLC indicating that, based upon the closing bid price of our Common Stock for the 30
consecutive business days between February 27, 2024 and April 9, 2024, we did not meet the minimum bid price of $1.00 per share required
for continued listing on Nasdaq pursuant to Nasdaq Listing Rule 5550(a)(2) (the “Bid Price Rule”). The Letter also indicated
that we were provided with a compliance period of 180 calendar days, or until October 7, 2024, in which to regain compliance with the
Bid Price Rule pursuant to Nasdaq Listing Rule 5810(c)(3)(A). We did not regain compliance with the Bid Price Rule by October 7, 2024,
and on October 8, 2024, Nasdaq notified us that our securities were subject to delisting from Nasdaq unless we timely requested a hearing
before the Nasdaq Hearings Panel (the “Panel”). We subsequently timely requested a hearing before the Panel, which was held
on December 5, 2024 (the “Hearing”).
On
November 19, 2024, we received an additional deficiency notice from the Staff indicating that we no longer satisfied the $2.5 million
stockholders’ equity requirement set forth in Nasdaq Listing Rule 5550(b)(1) (the “Equity Rule”) for continued listing
on Nasdaq. The Staff indicated that our non-compliance with the Equity Rule would be considered by the Panel at the Hearing and could
serve as an additional basis for delisting of our securities from Nasdaq.
On
December 26, 2024, we received a decision letter (the “Decision Letter”) from the Panel granting a limited extension of time
for us to demonstrate compliance with the Bid Price Rule and the Equity Rule for continued listing on Nasdaq, subject to the following
conditions: (i) on or before February 27, 2025, we will have obtained stockholder approval to effect a reverse stock split; (ii) on or
before March 31, 2025, we shall have effected a reverse stock split and, thereafter, maintain a $1.00 closing bid price of our common
stock for a minimum of ten consecutive trading days; (iii) on or before March 31, 2025, we are required to demonstrate compliance with
the Equity Rule by filing public disclosure with the SEC and demonstrate long-term compliance with the Equity Rule; and (iv) on or before
March 31, 2025, we are required to demonstrate compliance with all continued listing requirements for Nasdaq. On February 24, 2025, we
obtained approval from our stockholders to file a certificate of amendment to our Certificate of Incorporation to effectuate the Reverse
Stock Split, among others, and on March 13, 2025, the Reverse Stock Split became effective.
On
April 9, 2025, we received a letter (the “April Letter”) from the Staff notifying us that we had demonstrated compliance
with the Bid Price Rule and the Equity Rule as required by the Panel pursuant to the Decision Letter.
Pursuant
to the April Letter, we are subject to a mandatory panel monitor for a period of one year from the date of the April Letter. If, within
that one-year monitoring period, Staff finds us again out of compliance with the Equity Rule that was subject of the exception, notwithstanding
Rule 5810(c)(2), we will not be permitted to provide the Staff with a plan of compliance with respect to that deficiency and Staff will
not be permitted to grant additional time for us to regain compliance with respect to that deficiency, nor will the company be afforded
an applicable cure or compliance period pursuant to Rule 5810(c)(3). Instead, Staff will issue a Delist Determination Letter and we will
have an opportunity to request a new hearing with the Panel or a newly convened Hearings Panel if the initial Panel is unavailable.
Critical
Accounting Policies and Significant Estimates
This
management’s discussion and analysis of financial condition and results of operations is based on our financial statements, which
have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions
that affect the reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial
statements, and the reported amounts of revenue and expenses during the reported period. In accordance with U.S. GAAP, we base our estimates
on historical experience and on various other assumptions we believe to be reasonable under the circumstances. Actual results may differ
from these estimates if conditions differ from our assumptions. While our significant accounting policies are more fully described in
Note 3 – Summary of Significant Accounting Policies in the “Notes to Financial Statements”, we believe the following
accounting policies are critical to the process of making significant estimates in preparation of our financial statements.
Inventory
Inventories
are stated at the lower of cost or net realizable value. Net realizable value is the estimated selling prices in the ordinary course
of business, less reasonably predictable costs of completion, disposal, and transportation. Cost is determined using the “first-in,
first-out” method.
Inventory
write-offs are provided to cover risks arising from slow-moving items and obsolete items. The Company periodically evaluates the quantities
on hand relative to current and historical selling prices and historical and projected sales volume. Based on this evaluation, provisions
are made when required to write-down inventory to its net market value.
Impairment
of long-lived assets
The long-lived assets of the
Company, including finite-lived intangible assets, are reviewed for impairment in accordance with ASC No. 360, “Property, Plant
and Equipment”, whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable.
The recoverability of assets to be held and used is measured by a comparison of the carrying amount of an assets to the future undiscounted
cash flows expected to be generated by the assets. If such review indicates that the carrying amount of long-lived assets is not recoverable,
the carrying amount of such assets is reduced to fair value.
During the years ended December 31, 2025 and 2024, the Company recorded
a long-lived asset impairment charge of $645 and $0, respectively. See Note 21
Goodwill
impairment
Management
also evaluates goodwill for impairment at least annually, or more frequently if events or changes in circumstances indicate that the
carrying value may not be recoverable. Any impairment loss is recognized in the Consolidated Statements of Operations.
Business combination
The Company applies the provisions
of ASC 805, “Business Combination” and allocates the fair value of purchase consideration to the tangible assets acquired,
liabilities assumed, and intangible assets acquired based on their estimated fair values. The excess of the fair value of purchase consideration
over the fair values of these identifiable assets and liabilities is recorded as goodwill. Goodwill generated from a business combination
is primarily attributable to synergies.
When determining the fair values
of assets acquired and liabilities assumed, management makes significant estimates and assumptions, especially with respect to intangible
assets. Significant estimates in valuing certain intangible assets include but are not limited to future expected cash flows from acquired
technology and acquired customer relationships from a market participant perspective, useful lives and discount rates. Management’s
estimates of fair value are based upon assumptions believed to be reasonable, but which are inherently uncertain and unpredictable and,
as a result, actual results may differ from estimates. During the measurement period, which may extend up to one year from the acquisition
date, the Company may record adjustments to the provisional fair values of the assets acquired and liabilities assumed, with a corresponding
offset to goodwill. Upon the earlier of the end of the measurement period or the final determination of the fair values of the assets
acquired and liabilities assumed, any subsequent adjustments are recorded in earnings.
Acquisition-related expenses are recognized
separately from the business combination and are expensed as incurred. See Note 4 Merger, for further information.
Warrants
The
Company accounts for warrants as either equity-classified or liability-classified instruments based on an assessment of the
warrant’s specific terms and applicable authoritative guidance. The assessment considers whether the warrants are freestanding
financial instruments, meet the definition of a liability under ASC 480, and meet all of the requirements for equity classification,
including whether the warrants are indexed to the Company’s own common stock and whether the warrants meet the required
conditions for equity classification. This assessment, which requires the use of professional judgment, is conducted at the time of
warrant issuance and as of each subsequent reporting period end date while the warrants are outstanding. Warrants that meet all the
criteria for equity classification, are required to be recorded as a component of additional paid-in capital. Warrants that do not
meet all the criteria for equity classification, are required to be recorded as liabilities at their initial fair value on the date
of issuance and remeasured to fair value at each balance sheet date thereafter. The liability-classified warrants are recorded under
non-current liabilities. Changes in the estimated fair value of the warrants are recognized in Financial expenses or income in the
unaudited interim condensed consolidated statements of operations.
Revenue
recognition
Revenues
from product and services are recognized in accordance with ASC 606 “Revenue Recognition.” Five basic steps must be followed
before revenue can be recognized; (1) Identifying the contract(s) with a customer that create(s) enforceable rights and obligations;
(2) Identifying the performance obligations in the contract, such as promising to transfer goods or services to a customer; (3) Determining
the transaction price, meaning the amount of consideration in a contract to which an entity expects to be entitled in exchange for transferring
promised goods or services to a customer; (4) Allocating the transaction price to the performance obligations in the contract, which
requires the company to allocate the transaction price to each performance obligation on the basis of the relative standalone selling
prices of each distinct good or services promised in the contract; and (5) Recognizing revenue when (or as) the entity satisfies a performance
obligation by transferring a promised good or service to a customer.
The
Company’s performance obligation is generally the sale and delivery of its products. Revenues from product sales is recorded at
the net sales price, or “transaction price,” which includes estimates of variable consideration that result from discounts
as well as allowances for returns. Revenue from product sales is recognized at a point in time when control of the product is transferred,
which is generally upon shipment to the customer.
Regarding
its ENvue sales, the Company regularly sells its Systems and Nasoenteral tubes on a stand-alone basis and therefore concludes these products
are separate performance obligations. Revenue from product sales is recognized at a point in time when control of the product is transferred,
which is generally upon shipment to the customer.
When a
contract includes a combination of products and services, the transaction price is allocated to each performance obligation on a stand-alone
selling price basis. The stand-alone selling prices are generally determined based on the prices at which the Company separately sells
the products and services.
The
Company’s contracts with its ENvue customers generally do not include rights of return.
For
customers of both NanoVibronix and ENvue, payments are typically due between 30 and 60 days.
The
Company applied the practical expedient in ASC 606 and did not evaluate payment terms of one year or less for the existence of a significant
financing component. The related revenue is recognized net of any taxes collected from customers which are subsequently remitted to governmental
entities (e.g., sales tax and other indirect taxes). The Company elected to not disclose information about the remaining performance
obligations that have original expected durations of one year or less.
In
some of its contracts, the Company provides assurance type warranty services to its customers, in accordance with legal provisions or
industry standards to ensure the quality of the products. As such, the Company recognizes a provision for warranties in its financial
statements as applicable.
Income
taxes
We
account for income taxes in accordance with ASC 740, “Income Taxes”. This topic prescribes the use of the liability method
whereby deferred tax assets and liability account balances are determined based on differences between financial reporting and tax bases
of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences are expected
to reverse. We provide full valuation allowance, to reduce deferred tax assets to the amount that is more likely than not to be realized.
We
implemented a two-step approach to recognize and measure uncertain tax positions. The first step is to evaluate the tax position taken
or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than not
that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related appeals
or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% (cumulative basis)
likely to be realized upon ultimate settlement.
We
recognize interest and penalties related to uncertain tax positions on the income tax expense line in the accompanying consolidated statement
of operations. Accrued interest and penalties are included on the related tax liability line in the consolidated balance sheet.
Recently adopted accounting standards
In
December 2023, the Financial Accounting Standard Board (“FASB”) issued ASU 2023-09, Income Taxes (Topic 740),
Improvements to Income Tax Disclosures, which requires disaggregated information about the effective tax rate reconciliation as
well as information on income taxes paid. The guidance is effective for the Company for annual periods beginning January 1, 2025.
The Company adopted this guidance for the year ended December 31, 2025 on a prospective basis.
Recently issued accounting standards
In
November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosure
(Subtopic 220-40), Disaggregation of Income Statement Expenses, which requires disclosure of disaggregated information about certain
expense captions presented in the Consolidated Statements of Operations as well as disclosure about selling expense. The guidance will
be effective for the Company for annual periods beginning January 1, 2027 and interim periods beginning January 1, 2028, with early adoption
permitted. It could be applied either prospectively or retrospectively. The Company is currently evaluating the impact on its financial
statement disclosures.
In
July 2025, the FASB issued Accounting Standards Update No. 2025-05, “Financial Instruments — Credit Losses (Topic 326): Measurement
of Credit Losses for Accounts Receivable and Contract Assets” (“ASU 2025-05”), which provides a practical expedient
for estimating expected credit losses for current accounts receivable and current contract assets. ASU 2025-05 will be effective for
annual periods beginning after December 15, 2025 and interim periods within those annual reporting periods and should be applied prospectively.
The Company is currently evaluating the impact of ASU 2025-05 on its consolidated financial statements and related disclosures.
In September 2025, the FASB
issued ASU 2025-06, Intangibles - Goodwill and Other - Internal-Use Software (Subtopic 350-40):Targeted Improvements to the Accounting
for Internal-Use Software, to modernize the accounting for costs related to internal-use software and better align it with current software
development practices. The amended guidance removes references to project stages and clarifies when entities are required to begin capitalizing
eligible costs. This guidance will be effective for the Company for annual periods beginning January 1, 2028, with early adoption permitted.
The guidance may be applied prospectively, retrospectively, or using a modified prospective transition method. The Company is currently
evaluating the impact of this ASU on its consolidated financial statements.
In May 2025, the FASB issued ASU 2025-03, Business Combinations (Topic 805) and Consolidation (Topic 810): Determining
the Accounting Acquirer in the Acquisition of a Variable Interest Entity, which clarifies current guidance for determining the accounting
acquirer for a transaction effected primarily by exchanging equity interests in which the legal acquiree is a variable interest entity
that meets the definition of a business. ASU 2025-03 is effective for annual reporting periods beginning after December 15, 2026, including
interim reporting periods within those annual reporting periods, with early adoption permitted. The Company is currently evaluating the
impact of this ASU on its consolidated financial statements.
Results
of Operations
Year
Ended December 31, 2025, Compared to Year Ended December 31, 2024
Revenues. For the years
ended December 31, 2025 and 2024, revenues were approximately $2,553 and $2,558, respectively, representing a decrease of approximately
$5, or 0.2%, year over year. The decrease was primarily attributable to the removal of PainShield Ultra from the market, partially offset
by revenues generated from ENvue systems, as well as continued revenues from Veterans Administration facilities and workers’ compensation
programs and sales through certain sales representatives and our largest direct medical equipment distributor. Revenues may fluctuate
as new customers are added or when existing distributors or customers place large orders in one period and not in another.
For
the year ended December 31, 2025, the percentage of revenues attributable to our products was: PainShield and monthly kits 73%–
Envue system and tubes 27%. For the year ended December 31, 2024, the percentage of revenues attributable to our products was: PainShield
and monthly kits – 99% and UroShield – 1%. For the years ended December 31, 2025, and 2024, the portion of our revenues that
was derived from our largest direct medical equipment distributor, were 31% and 36%, respectively.
Gross
Profit. For the years ended December 31, 2025, and 2024, gross profit was approximately
$153 and $1,508, respectively, a decrease of approximately 90% or $1,355. Gross margin was also significantly impacted, declining primarily due to the removal of PainShield Ultra from the
market, which historically generated higher margins, as well as inventory write-downs associated with PainShield Ultra. In addition, amortization
expense related to intangible assets recognized in connection with the ENvue merger, which is recorded within cost of goods sold, further
contributed to the decline in gross margin.
Gross
profit as a percentage of revenues were approximately 6% and 59% for the years ended December 31, 2025, and 2024, respectively. The
increase in gross profit as a percentage of revenues is mainly due to the reasons described above.
Research
and Development Expenses. For the years ended December 31, 2025 and 2024, research and development expenses were approximately $1,762
and $909, respectively, representing an increase of approximately $853, or 94%. The increase was primarily attributable to additional
costs related to new intellectual property and patent activities, increased quality assurance and regulatory audit efforts associated
with the ENvue system, along with continued expenses related to product development activities.
Research
and development expenses as a percentage of total revenues were approximately 69% and 36% for the years ended December 31, 2025, and
2024, respectively.
Our
research and development expenses consist mainly of expenses related to subcontracting research and development and clinical trial activities,
as well as payroll expenses to employees, and the associated facilities’ costs, who are involved with research and development
activities.
Selling
and Marketing Expenses. For the years ended December 31, 2025 and 2024, selling and marketing expenses were approximately $2,493
and $720, respectively, representing an increase of approximately $1,773, or 246%. The increase was primarily attributable to
amortization expense related to intangible assets recognized in connection with the purchase price allocation, as well as higher
consulting fees and travel costs associated with system reactivation, new installations, commercialization activities, and
rebranding efforts for the ENvue system.
Selling
and marketing expenses as a percentage of total revenues were approximately 98% and 28% for the years ended December 31, 2025, and 2024,
respectively.
Selling
and marketing expenses consist mainly of payroll expenses to direct sales and marketing employees,
travel expenses, conventions, advertising and marketing expenses, rent and facilities expenses associated with and allocated to selling
and marketing activities.
General
and Administrative Expenses. For the years ended December 31, 2025 and 2024, general and administrative expenses were approximately $7,633 and
$3,461, respectively, representing an increase of approximately $4,172, or 121%. The increase was primarily attributable to higher legal
fees related to securities matters, merger-related expenses, and litigation, as well as increased accounting and professional consulting
fees incurred in connection with the business combination, severance costs associated with former executive management and termination costs with former board members.
Our
general and administrative expenses consist mainly of payroll expenses for management and administrative employees, stock-based compensation
expenses, accounting, legal and facilities expenses associated with general and administrative activities and costs associated with being
a publicly traded company.
General
and administrative expenses as a percentage of total revenues were approximately 299% and 135% for the years ended December 31, 2025,
and 2024, respectively.
Goodwill
and Intangible Assets Impairment Expense. For the year ended December 31, 2025, we recognized a non-cash impairment charge of approximately
$11,154 related to goodwill and intangible assets associated with the ENvue reporting unit, consisting of approximately $10,509 related
to goodwill and $645 related to intangible assets. The impairment resulted from our annual impairment assessment, during which the estimated
fair value of the reporting unit and certain long-lived assets was determined to be below their respective carrying values.
Financial
income (expense). For the years ended December 31, 2025, and 2024, financial income (expense), net was income of approximately $4,397 and expense of approximately $(104), respectively. This was primarily driven by a gain of approximately $6,204 related to the change in fair value of warrant liabilities
that was partially offset by warrant liability issuance expenses of $1,418, and interest expense
of $288 related to the Company’s loan.
Income
tax expense. For the year ended December 31, 2025, our income tax benefit was approximately $307. For the year ended December
31, 2024, our income tax expense was approximately $19. For 2025 and 2024 we recorded tax expense at our Israeli subsidiary based on
the appropriate tax rate and in addition for 2025 we recorded a change in the valuation allowance
Net
Loss. Our net loss increased by approximately $14,480 or 391%, to approximately $18,185 for the year ended December 31, 2025, from
approximately $3,705 during the same period in 2024. The increase in net loss resulted primarily from the factors described above.
Liquidity
and Capital Resources
We have incurred net losses of approximately $18,185 during the year ended
December 31, 2025. We also had negative cash flow from operating activities of $9,372 for the year ended December 31, 2025. Although we
had a cash balance of just over $4,254 as of December 31, 2025, we expect to continue to incur losses and negative cash flows from operating
activities, and therefore, we do not have sufficient resources to fund our operation for the next twelve months from the date of this
filing causing us to have substantial doubt of our ability to continue as a going concern. We will need to continue to raise additional
capital to finance its losses and negative cash flows from operations beyond the next years and may continue to be dependent on additional
capital raising as long as our products do not reach commercial profitability.
During the year ended
December 31, 2025, we met our short-term liquidity requirements from our existing cash reserves and equity financings. Our future capital requirements
and the adequacy of our available funds will depend on many factors, including our ability to successfully commercialize our
products, advance development of new products, and respond to evolving technological and market conditions. We expect to continue to
incur losses and negative cash flows from operations.
We intend to fund our operations through a combination of equity financings and potential strategic alliances with
third parties. However, there can be no assurance that we will be able to raise additional capital, when needed, on terms favorable to
us, or at all.
We
do not have any material commitments to capital expenditures as of December 31, 2025, and we are not aware of any material trends in
capital resources that would impact our business.
As
of December 31, 2025, we have no off-balance sheet transactions, arrangements, obligations (including contingent obligations), or other
relationships with unconsolidated entities or other persons that have, or may have, a material effect on our financial condition, changes
in financial condition, revenues or expenses, results of operations, liquidity, capital expenditures or capital resources.
September 2025 Registered Direct Offering
On
September 16, 2025, the Company entered into a securities purchase agreement with a single institutional investor, pursuant to which
the Company agreed to issue and sell (i) 74,114 shares of common stock, and (ii) pre-funded warrants to purchase up to 217,090 shares
of common stock in a registered direct offering for $2.04 million, pursuant to an effective shelf registration statement on Form S-3
(the “September 2025 Registered Direct Offering”). The offering price was $7.01 per share of common stock and $7.009 per
pre-funded warrant, which was the price of each share of common stock sold, minus the nominal $0.001 exercise price per pre-funded warrant.
The net proceeds from the September 2025 Registered Direct Offering were approximately $1.88 million, after deducting placement agent
fees of $163.
July
2025 Private Placement of Series H Preferred Stock
On
July 18, 2025, we entered into a Securities Purchase Agreement (the “Series H Purchase Agreement”) with a certain institutional
investor (the “Series H Investor”), pursuant to which we agreed to sell to the Series H Investor (i) an aggregate of 8,889
shares of our newly-designated Series H Convertible Preferred Stock, with a par value of $0.001 per share and a stated value of $1,000
per share, initially convertible into up to 880,099 shares of our common stock, at an initial conversion price of $10.10 per share (the
“Series H Preferred Stock”) pursuant to the Certificate of Preferences, Rights and Limitations of the Series H Convertible
Preferred Stock (the “Series H Certificate of Designations”) and (ii) warrants to acquire up to an aggregate of 467,836 shares
of common stock (the “Series H Warrants”) at an exercise price of $22.50 (such closing, the “July 2025 Initial Closing”).
Pursuant
to the terms of the Series H Purchase Agreement, we also agreed to issued 2,222 shares of Series H Preferred Stock with a total
stated value of $2,222 in a second closing and warrants to acquire up to an aggregate of 116,960 shares of common stock the Series H Warrants at an exercise
price of $22.50 (such closing, the “Series H Second Closing”), subject to the satisfaction of customary closing conditions. Additionally, pursuant to
the terms of the Series H Purchase Agreement, we have agreed that during the period ending 36 months from the effective date of the
registration statement (the “Series H Resale Registration Statement”) registering the resale of the shares of common
stock underlying the Series H Preferred Stock and the Series H Warrants, the Series H Investor shall have the right, but no
obligation, upon notice to us from time to time, to purchase up to an aggregate of $44,000 stated value (representing 44,000 shares
of Series H Preferred Stock and $39,600 of subscription amount) of additional Series H Preferred Stock.
The
Series H Initial Closing occurred on July 22, 2025, and the Series H Second Closing of the private placement occurred October 30, 2025. The
aggregate net proceeds from the Private Placement of Series H Preferred Stock was $9 million, after deducting placement agent fees and other offering
expenses payable by the Company of $958.
On January 30, 2026, we entered into that certain
Amendment Agreement (the “Series H Amendment Agreement”) with the Required Holders (as defined in the Series H Amendment Agreement).
Pursuant to the Series H Amendment Agreement, the Required Holders agreed to amend the Series H Certificate of Designations by filing
a Certificate of Amendment (the “Series H Certificate of Amendment”) to the Series H Certificate of Designations with the
Secretary of State of the State of Delaware to remove the Floor Price (as defined in the Series H Certificate of Designations) in consideration
of the holders of the Series H Preferred Stock exercising $2,500,000 of the Additional Investment Right (as such concept is described
in the Series H Purchase Agreement by and between us and the holders of the Series H Preferred Stock.
May
2025 Underwritten Public Offering, Series G Convertible Preferred Stock
On
May 16, 2025, we announced the closing of an underwritten public offering (the “2025 Underwritten Offering”) of 40,000
shares of our Series G Convertible Preferred Stock (“Series G Preferred Stock”), with a par value $0.01 per share and a
stated value equal to $25, and warrants to purchase up to 490,198 shares of common stock, par value $0.01 per share at an exercise
price of $20.40 per share (the “May 2025 Warrants”). The combined public offering price of each share of Series G
Preferred Stock together with an accompanying May 2025 Warrant was $25. The May 2025 Warrants have a term of five years from the
initial issuance date and are exercisable immediately upon issuance. We also issued to Dawson James Securities, Inc. warrants (the
“May 2025 Representative’s Warrants”) to purchase up to 24,510 shares of common stock the May 2025
Representative’s Warrants expire five years from the date of commencement of sales in the 2025 Underwritten
Offering.
Pursuant
to the terms of the Series G Certificate of Designations, the holders of the Series G Preferred Stock are entitled to receive cumulative
dividends at the rate per share of 9% per annum of the stated value per share until the fifth anniversary of the date of issuance of
the Series G Preferred Stock, which such dividends may be paid, at our option, in up to an aggregate of 220,588 shares of common stock.
The Series G holder may convert at any time and receive the full amount of dividends.
The
aggregate net proceeds of the 2025 Underwritten Offering were approximately $8.2 million, after deducting approximately $1.8 million
of underwriting discounts, commissions and other offering costs and expenses.
April
2025 Promissory Note and Guaranty
On
April 11, 2025, we issued a promissory note (the “April Note”) to Alpha (“Alpha” in the principal amount of $360
(the “April Note Principal Amount”), together with all accrued interest thereon. The April Note has a maturity date of June
11, 2025 (the “April Note Maturity Date”) and on the April Note Maturity Date, the aggregate unpaid April Note Principal
Amount, all accrued and unpaid interest and all other amounts payable under the April Note shall be due and payable. The April Note bore
interest at an annual rate equal to 8.0% and was payable “in kind” by adding such accrued interest to the April Note Principal
Amount. On May 19, 2025, we paid the April Note in full using proceeds from the 2025 Underwritten Offering, pursuant to the terms of
the April Note.
Summary
of Cash Flow
General.
As of December 31, 2025, we had cash of approximately $4,241, net compared to approximately $752 as of December 31, 2024. We have historically
met our cash needs through a combination of issuance of equity, borrowing activities and sales. Our cash requirements are generally for
product development, research and development costs, marketing and sales activities, general and administrative costs, capital expenditures
and general working capital.
Cash used in our operating activities was approximately $9,372 for the
years ended December 31, 2025, and approximately $2,516 for the same period in 2024. The increase in net cash used in operating activities
of approximately $8,330 was primarily attributable to non-cash charges, including goodwill and intangible asset impairment, issuance costs
allocated to warrant liabilities, depreciation and amortization, and non-cash interest expense, partially offset by a gain related to
the change in fair value of warrant liabilities and a benefit from deferred taxes.
Cash
provided by (used in) our investing activities was approximately $148 and ($3) for the years ended December 31, 2025, and 2024, respectively,
from cash acquired in the ENvue Merger and purchases of fixed assets.
Cash provided by financing activities during the years ended December 31,
2025, and 2024, was approximately $12,786 and $1, respectively, which was primarily composed of net proceeds received from the issuance
of common stock and preferred stock and warrants. Our future capital requirements and the adequacy of available funds will depend on many
factors, including our ability to successfully commercialize our products, our development of future products and competing technological
and market developments.
Factors
That May Affect Future Operations
We believe that our future operating
results will continue to be subject to quarterly variability based on a number of factors, including the ordering patterns of our distributors,
the timing of regulatory approvals, the progress and implementation of clinical trials, and manufacturing efficiencies associated with
the adoption of new materials and equipment.
Our operating results may also
be impacted by geopolitical developments, including hostilities in Israel and the broader Middle East, which could disrupt trade, impact
our ability to ship products, or affect our operations and those of our partners. In addition, fluctuations in foreign currency exchange
rates, including a weakening of the Euro or strengthening of the New Israeli Shekel against the U.S. dollar, may adversely affect our
results of operations.
More broadly, macroeconomic conditions, including changes in reimbursement policies and healthcare spending in the
markets in which we operate, may impact customer demand for our products.
ITEM
7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not
applicable.
ITEM
8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
Our
consolidated financial statements and the relevant notes to those statements are attached to this report beginning on page F-1.
ITEM
9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM
9A. CONTROLS AND PROCEDURES.
Disclosure
Controls and Procedures.
The
Company maintains disclosure controls and procedures (as defined in Rule 13a-15(e) under the Exchange Act) that are designed to ensure
that information required to be disclosed in the Company’s Exchange Act reports is recorded, processed, summarized and reported
within the time periods specified in SEC rules and forms, and that such information is accumulated and communicated to the Company’s
management, including our Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required
disclosure.
Limitations
on Effectiveness of Controls and Procedures
In
designing and evaluating our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act),
management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance
of achieving the desired control objectives. In addition, the design of disclosure controls and procedures must reflect the fact that
there are resource constraints, and that management is required to apply judgment in evaluating the benefits of possible controls and
procedures relative to their costs.
Evaluation
of Disclosure Controls and Procedures
Under
the PCAOB standards, a control deficiency exists when the design or operation of a control does not allow management or employees, in
the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. A significant deficiency
is a deficiency, or a combination of deficiencies, in internal control over financial reporting that is less severe than a material weakness,
yet important enough to merit the attention by those responsible for oversight of the company’s financial reporting. A material
weakness is a deficiency, or combination of deficiencies, in internal control over financial reporting, such that there is a reasonable
possibility that a material misstatement of the company’s annual or interim financial statements will not be prevented or detected
on a timely basis.
Under
the supervision and with the participation of our management, including our principal executive officer and principal financial officer,
we conducted an evaluation of our disclosure controls and procedures, as such term is defined under Rule 13a-15(e) and Rule 15d-15(e)
promulgated under the Exchange Act. Our management including the Chief Executive Officer and Chief Financial Officer has determined that,
as of December 31, 2025, the Company’s disclosure controls and procedures are ineffective and has concluded the consolidated financial
statements for the periods covered by and included in this Annual Report fairly present, in all material respects, our financial position,
results of operations and cash flows for the periods presented in conformity with U.S. GAAP.
Management’s
Report on Internal Control Over Financial Reporting
Our
management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over
financial reporting is defined in Rule 13a-15(f) and Rule 15d-15(f) under the Exchange Act as a process designed by, or under the supervision
of, the Company’s principal executive and principal financial officers and effected by the company’s board of directors,
management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation
of financial statements for external purposes in accordance with U.S. GAAP. Internal control over financial reporting includes policies
and procedures that:
| |
1) |
Pertain
to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the
assets of the Company; |
| |
|
|
| |
2) |
Provide
reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with
U.S. generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance
with authorizations of management and directors of the Company; and |
| |
|
|
| |
3) |
Provide
reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the Company’s
assets that could have a material effect on the financial statements. |
Because
of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of
any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions,
or that the degree of compliance with policies and procedures may deteriorate.
With
the participation of the Chief Executive Officer and Chief Financial Officer, our management conducted an evaluation of the effectiveness
of our internal control over financial reporting as of December 31, 2025 based on the criteria set forth by the Committee of Sponsoring
Organizations of the Treadway Commission, known as COSO, in Internal Control — Integrated Framework (2013).
Based
on this evaluation, our management, including the Chief Executive Officer and Chief Financial Officer, has concluded that our internal
control over financial reporting was ineffective as of December 31, 2025, as a result of the material weakness described below.
A
material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that
there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or
detected on a timely basis. In previously filed Annual Reports on Form 10-K’s, we disclosed material weaknesses related to the
design and effectiveness of our internal control over financial reporting.
We
did not have adequate controls in place to ensure adequate review, including the controls over managements review procedures for
processing, recording and reviewing transactions related to certain contracts, accounting memos and certain monthly closing
procedures. In addition, we identified deficiencies in certain aspects of our information technology general controls (“ITGCs”),
primarily related to the monitoring of our ERP systems and related processes, which impacted the effectiveness of certain automated controls
and management review procedures.
As
a smaller reporting company, the Company is not required to include in this Annual Report on Form 10-K a report on the effectiveness
of internal control over financial reporting by the Company’s independent registered public accounting firm.
Management’s
Remediation Plans
To
date, we have implemented certain measures to address the identified material weakness. These measures include increasing the use of
an accounting firm to provide and enhance our financial reporting and reviewing our closing procedures as well as improving our internal
controls. We intend to continue to take steps to remediate the material weakness described above and further evolve our internal controls
and processes. We will not be able to remediate these material weaknesses until these steps have been completed and have been operating
effectively for a sufficient period of time.
The
following remedial actions were taken through the year ended December 31, 2025:
| |
● |
With
assistance from a current finance and accounting third-party service provider, the Company was able to formalize our risk assessment
process, policies and procedures, implementing revised control activities, controls documentation, and ongoing monitoring activities
related to the internal controls over financial reporting including testing documentation to provide evidence that our system of
internal controls over financial reporting meets the requirements of the COSO 2013 framework, and provide a foundation for the Company
to communicate internal control deficiencies in a timely manner to those parties responsible for taking corrective action. |
| |
|
|
| |
● |
Expanded
consultations with third party specialists on complex accounting matters, financial reporting and regulatory filings, and create
enhanced documentation to support a more precise review process, as well as enhanced monitoring of the review process, and effective
enhanced monitoring of the review process, and an effective system of training of use and review of our inventory recording systems.
|
During
the period covered by this Annual Report on Form 10-K, we have not been able to remediate the material weaknesses identified above. Although
the Company has taken numerous steps, our remediation plan is not complete because we did not have adequate controls in place to ensure
adequate review, including the controls over managements review procedures for processing, recording and reviewing transactions related
to certain contracts, accounting memos and certain monthly closing procedures, and our remediation plan has not operated for a sufficient
period of time for the Company to complete testing to conclude that our newly implemented controls and procedures were operating effectively
as of December 31, 2025.
We
plan to enhance our testing plans and improve procedures to implement and maintain adequate controls over our financial processes and
reporting in the future, and maintain a system of testing to ensure our controls, procedures and management are operating effectively.
To address these internal control deficiencies, management will continue to perform additional analyses and other procedures to ensure
that the financial statements included herein fairly present, in all material respects, our financial position, results of operations
and cash flows for the periods presented.
In
addition, under the direction of the audit committee of the Board of Directors, management will continue to review and make necessary
changes to the overall design of the Company’s internal control environment, as well as to refine policies and procedures to improve
the overall effectiveness of internal control over financial reporting of the Company.
Changes
in Internal Control over Financial Reporting
During
the year ended December 31, 2025, there were several changes in our internal control over financial reporting that management believes
has materially improved our internal controls over financial reporting. These implemented changes included, but not necessarily limited
to: (i) conduct of a comprehensive review of existing controls related to information technology and systems relevant to financial statement
preparation; (ii) establishment of a formalized written set of policies and procedures, including testing documentation, to ensure compliance
with the COSO 2013 framework and maintaining comprehensive documentation of all control procedures, policies, and testing documentation;
(iii) development and implementation of proper accounting and reconciliation procedures for tracking the number of securities issued;
(iv) development and formalization of appropriate IT policies, including segregation of duties and monitoring procedures; and (v) engagement
of third-party consultants with expertise in internal controls and regulatory compliance to provide guidance and assistance in enhancing
control frameworks and addressing deficiencies effectively. In additional to the foregoing, from time to time, we make changes to our
internal control over financial reporting that are intended to enhance its effectiveness, and which do not have a material effect on
our overall internal control over financial reporting.
ITEM
9B. OTHER INFORMATION
None.
ITEM
9C. DISCLOSURES REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not
applicable
PART
III
ITEM
10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Board
of Directors
The
following table sets forth the name, age and positions of each director as of March 31, 2026.
| Name |
|
Age |
|
Position
with the Company |
| Doron
Besser, M.D. |
|
56 |
|
Chief
Executive Officer and Director |
| David
Johnson (1)(2)(3) |
|
67 |
|
Chairman
of the Board |
| Zeev
Rotstein, M.D.(1)(3) |
|
74 |
|
Director |
| Allison
Burgett(1)(2)(3) |
|
46 |
|
Director |
| Nino
Pionati(1)(2) |
|
68 |
|
Director |
| (1) |
Current
member of Compensation Committee. |
| (2) |
Current
member of Audit Committee. |
| (3) |
Current
member of Nominating and Corporate Governance Committee. |
The
following sets forth biographical information and the qualifications and skills for each director:
Doron
Besser, M.D., Chief Executive Officer and Director. Dr. Besser serves as Chief Executive Officer and President of ENvue Medical,
Inc. and brings more than two decades of leadership experience in medical device innovation, strategic commercialization, and organizational
growth. Prior to joining ENvue Medical, Dr. Besser served as Chief Executive Officer of Angioslide Ltd., a company focused on the development
of angioplasty products, where he led the company from early-stage development through animal and human clinical trials, regulatory approvals,
and initial commercial market penetration in Europe and the United States. Dr. Besser previously served as Vice President of Clinical
and Marketing and Vice President of Business Development at superDimension Ltd., a developer of minimally invasive pulmonology technologies,
where he was part of the founding leadership team and played a key role in identifying market opportunities and developing the company’s
clinical and commercial strategy. In 2012, superDimension was acquired by Covidien for approximately $300 million. Throughout his career,
Dr. Besser has focused on identifying innovative medical technologies and advancing them through the product development lifecycle, including
regulatory clearance, commercialization, and international market expansion. Dr. Besser holds a Doctor of Medicine degree from Ludwig
Maximilian University of Munich. Dr. Besser’s experience in the healthcare industry provides him with the qualifications and expertise
to serve as a member of our Board of Directors.
Dave
Johnson, Chairman of the Board. Mr. Johnson is the Chairman of ENvue Medical, Inc. and a seasoned healthcare executive with more
than 30 years of experience in the medical technology and biotechnology industries. Mr. Johnson most recently served as Non-Executive
Chairman of Advanced Medical Balloons from January 2022 to September 2025, where he helped guide the company through its acquisition
by Stryker. Previously, he served as Executive Chairman of the Board of Enveric Biosciences and as Chairman of the Board of HyperMed
Imaging, Inc. Mr. Johnson also served as Chief Executive Officer and a member of the Board of Directors of Alliqua BioMedical and as
a member of the Board of Directors and Chairman of the Compensation Committee of OMNI Orthopedics prior to its acquisition by Corin
Orthopedics. Earlier in his career, Mr. Johnson held senior leadership roles at ConvaTec, including Chief Executive Officer and member
of the Board of Directors, and previously held leadership positions at Bristol-Myers Squibb and Zimmer Biomet. Mr. Johnson holds an undergraduate
degree in business marketing from the Northern Alberta Institute of Technology in Edmonton, Alberta, Canada, completed the INSEAD Advanced
Management Program in Fontainebleau, France, and is a Fellow of the Wharton School of the University of Pennsylvania. Mr. Johnson’s
extensive leadership experience in global medical technology companies provides him with the qualifications and expertise to serve as
Chairman of the Board and a member of our Board of Directors.
Zeev
Rotstein, M.D. Professor Rotstein serves as a member of the Board of Directors of ENvue Medical, Inc. and is an internationally
recognized cardiologist and expert in health management systems with decades of experience in healthcare leadership, consultancy, and
academia. He spent 36 years at Sheba Medical Center, where he served in multiple leadership roles including Deputy Director, Director
of the Acute Care Hospital, and Director General from 2004 to 2016. From 2016 to 2021, he served as Chief Executive Officer and Director
General of Hadassah Medical Center. Professor Rotstein graduated from the Sackler School of Medicine Tel Aviv University and received
a Master of Health Administration from the Leon Recanati Graduate School of Business Administration at Tel Aviv University. He has held
fellowships at the New York Department of Health, Tufts University, and the Johns Hopkins Bloomberg School of Public Health. Professor
Rotstein’s experience in healthcare leadership and health systems management provides him with the qualifications and expertise
to serve as a member of our Board of Directors.
Allison
Burgett, Director. Ms. Burgett serves as a member of the Board of Directors of ENvue Medical, Inc. and is a finance
executive with more than two decades of leadership experience at publicly held companies. She currently serves as Chief Financial Officer
of Eagle Next Holdings (NYSE American: UAVS), a public technology and drone solutions company formerly known as AgEagle Aerial Systems,
Inc., where she leads financial planning, forecasting, and capital structure management and has strengthened financial operations and
internal controls. Prior to joining Eagle Next Holdings, Ms. Burgett held senior finance leadership roles at Centene Corporation
(NYSE: CNC), overseeing strategic budgeting, value capture initiatives, and performance management across multiple business units. Earlier
in her career, she held finance leadership positions at Republic Services (NYSE: RSG) and Providence Service Corporation (NASDAQ: PRSC),
where she supported acquisitions, divestitures, and operational integration initiatives. Ms. Burgett holds a Master of Business
Administration from Boise State University and a Bachelor of Science in Accounting from the University of Phoenix. Her financial leadership
experience and expertise in capital markets and corporate operations provide her with the qualifications to serve as a member of our
Board of Directors.
Nino
Pionati, Director. Mr. Pionati serves as a member of the Board of Directors of ENvue Medical, Inc. and is a healthcare and medical
technology executive with more than three decades of experience in strategy, commercialization, and international market expansion. From
2018 to 2021, he served as Chief Commercial Officer of ACell Inc., where he led global commercial operations and helped expand the company’s
international presence prior to its acquisition by Integra LifeSciences. From 2015 to 2018, he served as Chief Strategy and Marketing
Officer of Alliqua BioMedical, overseeing corporate strategy, marketing, research and development, and reimbursement initiatives. Earlier
in his career, Mr. Pionati held senior leadership roles at Bayer HealthCare and ConvaTec, including President, Intercontinental and President,
Continence and Critical Care, and previously held marketing leadership positions at Johnson & Johnson. Mr. Pionati holds a Master
of Business Administration from the Katz School of Business and an undergraduate degree from Concordia University in Montreal, Canada.
His experience in global medical technology strategy and commercialization provides him with the qualifications to serve as a member
of our Board of Directors.
Executive
Officers
The
following table sets forth the names, ages and positions of our executive officers and certain significant employees as of March 31,
2026.
| Name |
|
Age |
|
Position |
| Doron
Besser |
|
56 |
|
Chief
Executive Officer and Director |
| Nicole
Fernandez-McGovern |
|
53 |
|
Interim
Chief Financial Officer |
| Rita
Silberberg |
|
50 |
|
Executive
Vice President of Finance and Chief Accounting Officer |
Please
see the biography of Dr. Besser above in the section “Board of Directors.”
Nicole
Feranndez-McGovern, Interim Chief Financial Officer. Ms. Fernandez-McGovern serves as Chief Financial Officer of ENvue Medical,
Inc. and is a financial executive with more than 25 years of experience in corporate finance, operations, and strategic leadership across
public and private companies. She most recently served as Chief Financial Officer of NLS Pharmaceutics Ltd. (NASDAQ: NLSP) from October
2024 through November 2025, where she led the company through its merger with Kadimastem Ltd., completed in October 2025, and supported
the transition to the combined company, NewCelX (NASDAQ: NCEL). From October 2023 through May 2024, she served as Interim Chief Financial
Officer of Hayden AI, where she supported capital raises totaling $115 million and the execution of a $700 million contract with the
Metropolitan Transportation Authority. From 2016 to 2023, Ms. Fernandez-McGovern served as Chief Financial Officer and Executive Vice
President of Operations of Eagle Next Holdings Inc. (NYSE American: UAVS), where she oversaw global financial, operational, and manufacturing
functions and served as Interim Chief Executive Officer during the company’s transition to a new CEO in May 2020. Previously, she
served as Chief Executive Officer and Chief Financial Officer of Trunity Holdings, Inc., where she led the company’s restructuring.
Earlier in her career, she held financial management roles at Elizabeth Arden and began her career in the audit and assurance practice
at KPMG LLP. Ms. Fernandez-McGovern has also served on the boards of several public and private companies, including MGO Global, Inc.
She holds a Bachelor of Business Administration and a Master of Business Administration from the University of Miami and has been a licensed
Certified Public Accountant in Florida since 1998.
Rita
Silberberg, Executive Vice President of Finance and Chief Accounting Officer. Ms. Silberberg serves as Chief Financial Officer
of ENvue Medical, Inc. and has more than 15 years of experience in corporate finance, international tax planning, and financial management.
Prior to joining ENvue Medical in 2022, she served as Chief Financial Officer of Gravity Creative Space and held finance leadership roles
including controller at novoGI and firm controller and project manager at Ernst & Young. Ms. Silberberg has extensive experience
with accounting and financial reporting standards, including U.S. GAAP and IFRS, and has worked with multinational organizations across
multiple jurisdictions. Ms. Silberberg holds a B.A. in Business specializing in Accounting and an LL.B. in Accounting and Finance from
the College of Management.
CORPORATE
GOVERNANCE
ENvue
Medical, Inc., with the oversight of the Board and its committees, operates within a comprehensive plan of corporate governance for the
purpose of defining independence, assigning responsibilities, setting high standards of professional and personal conduct and assuring
compliance with such responsibilities and standards. We regularly monitor developments in the area of corporate governance.
Code
of Business Conduct and Ethics
We
have adopted a code of business conduct and ethics that applies to all of our officers, directors and employees. The code of business
conduct and ethics addresses, among other things, competition and fair dealing, conflicts of interest, financial matters and external
reporting, our funds and assets, confidentiality and corporate opportunity requirements and the process for reporting violations of the
code of business conduct and ethics, employee misconduct, improper conflicts of interest or other violations. A copy of the code of ethics
was attached as Exhibit 14.1 to our Annual Report on Form 10-K for the fiscal year ended December 31, 2016, and filed with the Securities
and Exchange Commission on March 31, 2017. If we amend or grant a waiver of one or more of the provisions of our code of business conduct
and ethics, we intend to satisfy the requirements under Item 5.05 of Form 8-K regarding the disclosure of amendments to, or waivers from,
provisions of our code of business conduct and ethics that apply to our principal executive, financial and accounting officers by posting
the required information on our website at www.envuemed.com within four business days following the date of such amendment or
waiver.
Board
Composition
Our
Certificate of Incorporation and Bylaws provide that our Board will consist of such number of directors as determined from time to time
by resolution adopted by our Board. The size of our Board is currently fixed at eight directors. Subject to any rights applicable to
any then outstanding preferred stock, any vacancies or newly created directorships resulting from an increase in the authorized number
of directors may be filled by a majority of the directors then in office. Each member of our Board is elected for a one-year term and
is elected at each annual meeting of stockholders.
We
have no formal policy regarding Board diversity. Our Board believes that each director should have a basic understanding of our principal
operational and financial objectives and plans and strategies, our results of operations and financial condition and relative standing
in relation to our competitors. We take into consideration the overall composition and diversity of the Board and areas of expertise
that director nominees may be able to offer, including business experience, knowledge, abilities and customer relationships. Generally,
we will strive to assemble a Board that brings to us a variety of perspectives and skills derived from business and professional experience
as we may deem are in our and our stockholders’ best interests. In doing so, we will also consider candidates with appropriate
non-business backgrounds.
Director
Independence and Committee Qualifications
We
are currently listed on The Nasdaq Capital Market and therefore rely on the definition of independence set forth in the Nasdaq Listing
Rules (“Nasdaq Rules”). Under the Nasdaq Rules, a director only qualifies as an “independent director” if, in
the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise
of independent judgment in carrying out the responsibilities of a director.
In
order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other
than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly
or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated
person of the listed company or any of its subsidiaries.
Our
Board undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information
requested from and provided by each director concerning his or her background, employment and affiliations, including family relationships,
our Board has determined that each of David Johnson, Alison Burgett, Nino Pionati, and Zeev Rotstein, M.D., or four of our five
directors, do not have a relationship (other than being a director and/or a stockholder of the Company) that would interfere with the
exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent”
as that term is defined under the Nasdaq Rules.
Our
Board of Directors has determined that (i) Alison Burgett, David Johnson, Zeev Rotstein and Nino Pionati, who compose our Audit
Committee, (ii) Zeev Rotstein, David Johnson, Nino Pionati and Alison Burgett, who compose our Compensation Committee, and (iii)
David Johnson, Alison Burgett, Nino Pionati and Zeev Rotstein, who compose our Nominating and Corporate Governance Committee,
each satisfy the independence standards for those committees established by the applicable rules and regulations of the Securities and
Exchange Commission and the Nasdaq Stock Market rules. In making this determination, our Board considered the relationships that each
non-employee director has with the Company and all other facts and circumstances our Board deemed relevant in determining their independence,
including the beneficial ownership of our capital stock by each non-employee director. We intend to comply with all applicable committee
composition and independence requirements within the time periods prescribed by Nasdaq.
Board
Committees, Meetings and Attendance
During
the year ended December 31, 2025, the Board held 20 meetings. We expect our directors to attend Board meetings, meetings of any
committees and subcommittees on which they serve and each annual meeting of stockholders, either in person or by teleconference. During
the year ended December 31, 2025, each director attended at least 50% of the total number of meetings held by the Board and Board committees
of which such director was a member. Last year’s annual meeting was not attended by any directors.
Our
Board currently has three standing committees which consist of an audit committee (the “Audit Committee”), a nominating and
corporate governance committee (the “Nominating and Corporate Governance Committee”) and a compensation committee (the “Compensation
Committee”), each of which has the composition and responsibilities described below.
Each
of these committees operates under a charter that has been approved by our Board. The current charter of each of these committees is
available on our website at www.envuemed.com in the “Governance” section under “Investors.” The reference
to our website address does not constitute incorporation by reference of the information contained at or available through our website,
and you should not consider it to be a part of this proxy statement.
Audit
Committee. As of the date of this Annual Report on Form 10-K, the Audit Committee is comprised of Alison Burgett (Chair),
David Johnson, Nino Pionati, and Zeev Rotstein, M.D., each of whom our Board has determined to be financially literate and qualify as
an independent director under Sections 5605(a)(2) and 5605(c)(2) of the Nasdaq Rules and Rule 10A-3(b)(1) of the Securities Exchange
Act of 1934, as amended (the “Exchange Act”). In addition, Ms. Burgett qualifies as an “audit committee financial
expert,” as defined in Item 407(d)(5)(ii) of Regulation S-K. Our Board also determined that each member of our Audit Committee
can read and understand fundamental financial statements in accordance with applicable requirements. In arriving at these determinations,
the Board examined each Audit Committee member’s scope of experience and the nature of their employment in the corporate finance
sector.
The
function of the Audit Committee is to assist the Board in its oversight of (1) the integrity of our financial statements, (2) our compliance
with legal and regulatory requirements, (3) the qualifications, independence and performance of our independent auditors and (4) audit
and non-audit fees and services.
The
audit committee met 4 times during the year ended December 31, 2025.
Prior
to each of Mr. Ferguson, Thomas Mika, Christopher Fashek and Maria Schroeder’s resignation from the Board and all committees thereto
in 2025, each of Michael Ferguson, Thomas Mika, Christopher Fashek and Maria Schroeder were financially literate and qualified as independent
directors under Sections 5605(a)(2) and 5605(c)(2) of the Nasdaq Rules and Rule 10A-3(b)(1) of the Exchange Act.
Nominating
and Corporate Governance Committee. As of the date of this Annual Report on Form 10-K, the Nominating and Corporate Governance Committee
is comprised of David Johnson (Chair), Alison Burgett, and Zeev Rotstein, M.D., each of whom our Board has determined qualifies
as an independent director under Section 5605(a)(2) of the Nasdaq Rules.
The
primary function of the Nominating and Corporate Governance Committee is to identify individuals qualified to become board members, consistent
with criteria approved by the Board, and select the director nominees for election at each annual meeting of stockholders as well as
reviewing the Company’s corporate governance policies and any related matters.
The
Nominating and Corporate Governance Committee met 1 time during the year ended December 31, 2025.
Prior
to Martin Goldstein, Christopher Fashek, and Maria Schroeder’s resignation from the Board and all committees thereto in 2025, each
of Martin Goldstein, Christopher Fashek, and Maria Schroeder qualified as an independent director under Section 5604(a)(2) of the Nasdaq
Rules.
Compensation
Committee. The Compensation Committee is comprised of Thomas Mika and Ms. Aurora Cassirer, each of whom our Board has determined
qualifies as an independent director under Sections 5605(a)(2) and 5605(d)(2) of the Nasdaq Rules, as an “outside director”
for purposes of Section 162(m) of the Internal Revenue Code and as a “non-employee director” for purposes of Section 16b-3
under the Exchange Act. The function of the compensation committee will be to discharge the Board’s responsibilities relating to
compensation of our directors and executives and our overall compensation programs.
The
primary objective of the Compensation Committee will be to develop and implement compensation policies and plans that are appropriate
for us in light of all relevant circumstances and which provide incentives that further our long-term strategic plan and are consistent
with our culture and the overall goal of enhancing enduring stockholder value.
The
Compensation Committee met 1 times during the year ended December 31, 2025.
Prior
to Michael Ferguson, Thomas Mika, Christopher Fashek and Maria Schroer’s resignation from the Board and all committees thereto
in 2025, each of Thomas Mika, Christopher Fashek and Michael Ferguson qualified as an independent director under Sections 5605(a)(2)
and 5605(d)(2) of the Nasdaq Rules, as an “outside director” for purposes of Section 162(m) of the Internal Revenue Code
and as a “non-employee director” for purposes of Section 16b-3 under the Exchange Act.
Board
Leadership Structure
The
Board is committed to promoting our effective, independent governance. Our Board believes it is in our best interests and the best interests
of our stockholders for the Board to have the flexibility to select the best director to serve as chairman at any given time, regardless
of whether that director is an independent director or the chief executive officer. Consequently, we do not have a policy governing whether
the roles of chairman of the Board and chief executive officer should be separate or combined. This decision is made by our Board, based
on our best interests considering the circumstances at the time.
Currently,
the offices of the Chairman of the Board and the Chief Executive Officer are held by two different people. David Johnson is our independent,
non-executive chairman of the Board, and Doron Besser is our chief executive officer. The Chief Executive Officer will be responsible
for our day-to-day leadership and performance, while the chairman of the Board will provide guidance to the chief executive officer and
set the agenda for board meetings and preside over meetings of the Board. We believe that separation of the positions will reinforce
the independence of the Board in its oversight of our business and affairs, and create an environment that is more conducive to objective
evaluation and oversight of management’s performance, increasing management accountability and improving the ability of the Board
to monitor whether management’s actions are in our best interests and those of our stockholders.
Role
in Risk Oversight
Our
Board oversees an enterprise-wide approach to risk management, designed to support the achievement of business objectives, including
organizational and strategic objectives, to improve long-term organizational performance and enhance stockholder value. The involvement
of our Board in setting our business strategy is a key part of its assessment of management’s plans for risk management and its
determination of what constitutes an appropriate level of risk for the company. The participation of our Board in our risk oversight
process includes receiving regular reports from members of senior management on areas of material risk to our company, including operational,
financial, legal and regulatory, and strategic and reputational risks, including cybersecurity.
The
Board continually reviews the Company’s controls and procedures that involve cybersecurity matters to determine the potential material
impact to our financial results, operations, and/or reputation to ensure such incidents are immediately reported by management to the
Board, or individual members or committees thereof, as appropriate, in accordance with our escalation framework.
While
our Board has the ultimate responsibility for the risk management process, senior management and various committees of our Board will
also have responsibility for certain areas of risk management.
Our
senior management team is responsible for day-to-day risk management and regularly reports on risks to our full Board or a relevant committee.
Our finance and regulatory personnel serve as the primary monitoring and evaluation function for company-wide policies and procedures,
and manage the day-to-day oversight of the risk management strategy for our ongoing business. This oversight includes identifying, evaluating,
and addressing potential risks that may exist at the enterprise, strategic, financial, operational, compliance and reporting levels.
The
audit committee will focus on monitoring and discussing our major financial risk exposures and the steps management has taken to monitor
and control such exposures, including our risk assessment and risk management policies. As appropriate, the audit committee will provide
reports to and receive direction from the full Board regarding our risk management policies and guidelines, as well as the audit committee’s
risk oversight activities.
In
addition, the compensation committee will assess our compensation policies to confirm that the compensation policies and practices do
not encourage unnecessary risk taking. The compensation committee will review and discuss the relationship between risk management policies
and practices, corporate strategy and senior executive compensation and, when appropriate, report on the findings from the discussions
to our Board. Our compensation committee intends to set performance metrics that will create incentives for our senior executives that
encourage an appropriate level of risk-taking that is commensurate with our short-term and long-term strategies.
Communications
with Directors
The
Board welcomes communication from our stockholders. Stockholders and other interested parties who wish to communicate with a member or
members of our Board or a committee thereof may do so by addressing correspondence to the Board member, members or committee, c/o Envue
Medical, Inc., 969 Pruitt Place, Tyler TX 75703, Attn: Doron Besser, Chief Executive Officer. Our Chief Executive Officer will review
and forward correspondence to the appropriate person or persons.
All
communications received as set forth in the preceding paragraph will be opened by the Chief Executive Officer for the sole purpose of
determining whether the contents represent a message to our directors. Any contents that are not in the nature of advertising, promotions
of a product or service or patently offensive material will be forwarded promptly to the addressee(s). In the case of communications
to the Board or any group or committee of directors, the Chief Executive Officer will make sufficient copies of the contents to send
to each director who is a member of the group or committee to whom the communication is addressed. If the amount of correspondence received
through the foregoing process becomes excessive, our Board may consider approving a process for review, organization and screening of
the correspondence by the corporate secretary or another appropriate person.
Family
Relationships
There
are no family relationships among our directors and executive officers, or person nominated or chosen by the Company to become a director
or executive officer.
Involvement
in Certain Legal Proceedings
None
of our directors or executive officers has been involved in any of the following events during the past ten years:
| |
● |
any
bankruptcy petition filed by or against any business of which such person was a general partner or executive officer either at the
time of the bankruptcy or within two years prior to that time; |
| |
● |
any
conviction in a criminal proceeding or being subject to a pending criminal proceeding (excluding traffic violations and other minor
offences); |
| |
● |
being
subject to any order, judgment, or decree, not subsequently reversed, suspended or vacated, of any court of competent jurisdiction,
permanently or temporarily enjoining, barring, suspending or otherwise limiting his or her involvement in any type of business, securities
or banking activities; or |
| |
● |
being
found by a court of competent jurisdiction (in a civil action), the SEC or the Commodity Futures Trading Commission to have violated
a federal or state securities or commodities law, and the judgment has not been reversed, suspended, or vacated. |
Insider
Trading Policy and Anti-Hedging Policy
We
maintain an insider trading policy that applies to our officers and directors that prohibits trading our securities during certain established
periods and when in possession of material non-public information. It also prohibits, unless approved in advance in limited circumstances
by the policy administrator, the hedging of our securities, including short sales or purchases or sales of derivative securities based
on our securities, and the use of our securities to secure a margin or other loan. Since the adoption of our insider trading policy,
the policy administrator has not granted any such exemptions to the policy’s general prohibition on hedging or pledging. While
the Company is not subject to the insider trading policy, the company does not trade in its securities when it is in possession of material
nonpublic information other than pursuant to previously adopted Rule 10b5-1 trading plans.
Section
16(a) Beneficial Ownership Reporting Compliance
Section
16(a) of the Exchange Act requires that each of our directors and executive officers, and any other person who owns more than ten percent
(10%) of our common stock, file with the SEC initial reports of ownership and reports of changes in ownership of our common stock. To
our knowledge, based solely on information furnished to us and written representations by such persons that no such other reports were
required to be filed, we believe that all such SEC filing requirements were met in a timely manner during the year ended December 31,
2025, other than with respect to the following:
On
December 25, 2025, December 29, 2025, January 12, 2026, and February 26, 2026, Form 43s for each of David Johnson, Alison Burgett,
Zeev Rotstein and Nicole Fernandez-McGovern were filed late in connection with the commencement of such persons becoming directors or
officers, as applicable, of the Company.
ITEM
11. EXECUTIVE COMPENSATION
The
following table sets forth the names and positions of: (i) each person who served as our principal executive officer during the year
ended December 31, 2025, (ii) the two most highly compensated executive officers, other than our principal executive officer, who were
serving as executive officers, as determined in accordance with the rules and regulations promulgated by the SEC, as of December 31,
2025, (iii) up to two additional individuals for whom disclosure would have been provided pursuant to clause (ii) but for the fact that
the person was not serving as our executive officer at December 31, 2025 (collectively our “Named Executive Officers”):
| Name |
|
Position |
| Doron
Besser |
|
Chief
Executive Officer |
| Nicole
Fernandez-McGovern |
|
Interim
Chief Financial Officer |
| Brian
Murphy |
|
Former
Chief Executive Officer |
| Stephen
Brown |
|
Former
Chief Financial Officer |
Summary
Compensation Table
The
following table sets forth all compensation earned, in all capacities, during the fiscal years ended December 31, 2025, and 2024 by our
Named Executive Officers.
Name
and Principal Position | |
Year | | |
Salary
($) | | |
Bonus ($)(1) | | |
Option
Awards ($)(2) | | |
All
Other Compensation ($)(3) | | |
Total ($)(2) | |
| Doron Besser, Chief Executive Officer | |
| 2025 | | |
$ | 354,744 | | |
$ | 224,647 | | |
| - | | |
$ | 22,269 | | |
$ | 601,660 | |
| | |
| 2024 | | |
$ | - | | |
| - | | |
| - | | |
| - | | |
$ | - | |
| Nicole Fernandez-McGovern, Interim Chief Financial
Officer | |
| 2025 | | |
$ | 12,500 | | |
| - | | |
| - | | |
| - | | |
$ | 12,500 | |
| | |
| 2024 | | |
$ | - | | |
| - | | |
| - | | |
| - | | |
$ | - | |
| Brian Murphy, Former Chief Executive
Officer | |
| 2025 | | |
$ | 134,397 | | |
| 73,200 | | |
| - | | |
| 321,000 | | |
$ | 528,597 | |
| | |
| 2024 | | |
$ | 307,269 | | |
| 83,712 | | |
| 42,285 | | |
| - | | |
$ | 433,266 | |
| | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | |
| Stephen Brown, Former Chief Financial Officer | |
| 2025 | | |
$ | 293,465 | | |
| 94,500 | | |
| - | | |
| 180,000 | | |
$ | 567,965 | |
| | |
| 2024 | | |
$ | 255,385 | | |
| 44,500 | | |
| 26,428 | | |
| - | | |
$ | 326,313 | |
| (1) |
Represents
incentive compensation payments earned. |
| (2) |
In
accordance with SEC rules, the amounts reported in this column reflect the aggregate grant date fair value of stock option awards
granted during the applicable fiscal year, computed in accordance with ASC Topic 718, Compensation—Stock Compensation. The
fair value is based on the Black-Scholes option pricing model using the market price of the underlying shares at the grant date.
For additional discussion of the valuation assumptions used in determining stock-based compensation and the grant date fair value
for stock options, see “Management’s Discussion and Analysis of Financial Condition and Results of Operation - Critical
Accounting Policies - Stock-based compensation” and Note 3- “Summary of Significant Accounting Policies”
and Note 11- “Stockholders’ Equity (Deficiency)” to our audited consolidated financial statements for the fiscal
year ended December 31, 2025, included herein. |
| (3) |
Represents car allowance for Dr. Besser, and lump sum severance payments for Mr. Murphy in the amount of $321 and $180 for Mr. Brown per
their employment agreements. |
Narrative
Disclosure to Summary Compensation Table
Employment
Agreements
We
have entered into agreements with each of our Named Executive Officers. A description of each of these agreements follows.
Doron
Besser, Chief Executive Officer
2025
Besser Employment Agreement
On
December 17, 2025, ENvue Medical Israel Ltd., our wholly-owned subsidiary, entered into an Amended and Restated Employment Agreement
with Dr. Doron Besser, our Chief Executive Officer, which was subsequently amended by the First Amendment to the Amended and Restated
Employment Agreement, dated February 2, 2026 (collectively the “Besser Employment Agreement”). The following is a summary
of the material terms of the Besser Employment Agreement.
Pursuant
to the Besser Employment Agreement, Dr. Besser is entitled to a gross base salary of $360,000 less any applicable payroll deductions
and tax withholdings and pursuant to the exchange rate as set forth in the Besser Employment Agreement (the “Besser Base Salary”).
Additionally, Dr. Besser shall be eligible to receive a gross annual bonus of up to $180,000, less applicable payroll deductions and
tax withholdings, pursuant to the exchange rate as set forth in the Besser Employment Agreement, and subject to the extent to which Dr.
Besser has met performance criteria for the applicable calendar year and as determined by the Board of Directors.
Pursuant
to the Besser Employment Agreement, and upon the Board’s approval of such grants on April 4, 2026, we granted Dr. Besser
an initial award of restricted stock units (the “Initial Grant”) representing, in the aggregate, nine percent (9%) of
our issued and outstanding common stock on a fully diluted basis as of the date of grant, subject to the terms and conditions of our
2024 Long-Term Incentive Plan and a restricted stock unit award agreement. The Initial Grant is intended to be made, to the extent
possible, under the Capital Gains track of Section 102 of the Israeli Tax Ordinance and shall be fully vested at grant. In addition,
we shall issue additional restricted stock units to Dr. Besser on each quarterly anniversary of the date of the Initial Grant, to
the extent necessary to maintain his nine percent (9%) fully diluted ownership interest (each, a “Gross-Up”), provided
Dr. Besser remains employed on the applicable date of grant. Each Gross-Up shall also be fully vested at grant. In the event Dr.
Besser is terminated without Cause or resigns for Good Reason, he shall receive a prorated Gross-Up immediately prior to his
termination date, based on the number of calendar days employed during the applicable quarter. As of the date of this Annual Report
on Form 10-K, the grant date for the Initial Grant has not yet been determined.
Pursuant
to the terms of the Besser Employment Agreement, the Company shall transfer the funds each month to a study fund chosen by Dr. Besser
(the “Study Fund”) in the following amounts: (i) 2.5% of the Besser Base Salary, to be contributed by the Dr. Besser and
deducted from the Base Salary; and (b) a sum equal to 7.5% of the Besser Base Salary, to be contributed by the Company, all, up to the
maximum cap permitted by the Israeli tax authorities for exemption. Upon termination of Dr. Besser’s employment, the Company shall
remit to Dr. Besser all sums accumulated for Dr. Besser’s benefit in the Study Fund.
The
Besser Employment Agreement shall be at-will and either the Company or Dr. Besser may terminate Dr. Besser’s employment with the
Company at any time upon six months’ written notice to the other party, subject to the terms of the Besser Employment Agreement.
In the event Dr. Besser’s employment is terminated by the Company without Cause or Dr. Besser resigns for Good Reason, or if a
Change in Control of the Company has occurred, and Dr. Besser has resigned for Good Reason, or is terminated for reasons other than for
Cause (each as defined in the Besser Employment Agreement), within 90 days from the occurrence of such Change in Control of the Company
and provided that Dr. Besser executes and delivers a separation and release agreement in a form acceptable to the Company, within 21
days after Dr. Besser’s date of termination, subject to applicable law, among others, the Company shall pay the Dr. Besser a special
one-time gross payment equal to 12 months’ Besser Base Salary in effect on the date of termination), less any applicable payroll
deductions and tax withholdings. Dr. Besser shall also be entitled to severance pay in an amount equal to 81/3%
of the Besser Base Salary and certain pension and insurance contributions.
The
Besser Employment Agreement also provides for certain customary covenants regarding non-solicitation, non-competition and confidentiality
and certain customary employee benefits regarding transport and technology expenses, among others.
For
the years ended December 31, 2025, and December 31, 2024, the Compensation Committee approved performance bonuses of $224,647 and $0,
respectively to Dr. Besser.
Nicole
Fernandez-McGovern, Chief Financial Officer
2025
Fernandez-McGovern Employment Agreement
On
December 18, 2025, we entered into a consulting agreement (the “Fernandez-McGovern Consulting Agreement”) with RCM Financial
Consulting, Inc., through which Nicole Fernandez-McGovern serves as our interim Chief Financial Officer. Ms. Fernandez-McGovern is an
independent contractor and is not an employee of the Company.
Pursuant
to the terms of the Fernandez-McGovern Consulting Agreement, the Company shall pay Ms. Fernandez McGovern an annual salary of $300,000
as well as reimbursement of certain customary expenses. The Fernandez-McGovern Consulting Agreement may be terminated by either Ms. Fernandez-McGovern
or the Company at any time by providing 60 days prior written notice, pursuant to, and in accordance with, the terms of the Fernandez-McGovern
Consulting Agreement. Pursuant to the terms of the Fernandez-McGovern Consulting Agreement, Ms. McGovern shall perform customary tasks
for such a position, including but not limited to, overseeing and perform technical accounting review under GAAP and SEC requirements,
leading and conclude impairment testing, going concern analysis, and complex accounting judgments and maintaining responsibility for
the accuracy, completeness, and defensibility of the Company’s financial statements. The Fernandez-McGovern Consulting Agreement
also provides for certain customary provisions regarding confidentiality.
Brian
Murphy, Former Chief Executive Officer
On
June 4, 2025, the Board appointed Doron Besser, M.D., a director of the Board and Chief Executive Officer and President of the Company’s
wholly-owned subsidiary, ENvue Medical Holdings LLC, as the Company’s CEO, effective as of the same date. In connection with such
appointment, Brian Murphy resigned from the Company as its CEO, effective as of the same date. Prior to Mr. Murphy’s resignation
from the Company as its CEO, Mr. Murphy was employed by the Company pursuant to the 2024 Murphy Employment Agreement (as defined below).
2024
Murphy Employment Agreement
On
September 20, 2024, we entered into a new employment with Mr. Murphy (the “2024 Murphy Employment Agreement”), pursuant to
which the parties agreed to have Mr. Murphy continue to serve as our Chief Executive Officer, effective September 20, 2024, through August
31, 2025, unless earlier terminated by either party pursuant to the 2024 Murphy Employment Agreement. The prior Employment Agreement
by and between the Company and Mr. Murphy, dated as of January 1, 2022, terminated upon effectiveness of the 2024 Murphy Agreement.
As
consideration for his services as Chief Executive Officer, Mr. Murphy was entitled to receive (i) an annual base salary of $321,000,
less applicable payroll deductions and tax withholdings; (ii) reimbursement of any reasonable and customary, documented out-of-pocket
expenses actually incurred by Mr. Murphy in connection with the performance of his services under the 2024 Murphy Employment Agreement;
and (iii) an annual bonus of up to $100,000, less applicable payroll deductions and tax withholdings, based on the extent to which Mr.
Murphy met performance criteria for the calendar year, as determined by us in good faith. Mr. Murphy may also be eligible to receive
certain grants of incentive stock options to purchase shares of common stock.
Either
party was permitted to terminate the 2024 Murphy Employment Agreement at any time upon ninety (90) days written notice. Upon termination
of Mr. Murphy’s employment, we agreed to pay Mr. Murphy (i) any unpaid salary accrued through the date of termination, (ii) any
accrued and unpaid vacation or similar pay to which Mr. Murphy is entitled as a matter of law or Company policy, and (iii) any unreimbursed
expenses properly incurred prior to the date of termination (the “Murphy Accrued Obligations”).
In
the event we terminated Mr. Murphy’s employment for cause, we had no further liability or obligation to Mr. Murphy under the 2024
Murphy Employment Agreement or in connection with Mr. Murphy’s employment, except for the Murphy Accrued Obligations. The 2024
Murphy Employment Agreement also contained certain standard non-competition, non-solicitation, confidentiality, and assignment of inventions
requirements for Mr. Murphy.
For
the years ended December 31, 2025, and December 31, 2024, the Compensation Committee approved performance bonuses of $73,200 and $83,712,
respectively, to Mr. Murphy.
Stephen
Brown, Former Chief Financial Officer
On
December 18, 2025, the Board appointed Nicole Fernandez-McGovern, as the Company’s interim Chief Financial Officer, effective as
of the same date. In connection with such appointment, Stephen Brown resigned from the Company as its Chief Financial Officer, effective
as of the same date. Prior to Mr. Brown’s resignation from the Company, Mr. Murphy was employed by the Company pursuant to the
A&R Brown Employment Agreement (as defined below).
2025
Amended and Restated Brown Employment Agreement
On
August 11, 2025 (the “Brown Effective Date”), the Company entered into an amended and restated employment agreement (the
“A&R Brown Employment Agreement”) with Mr. Brown, which such agreement amends, restates and the employment agreements
dated September 20, 2024, January 1, 2022, and October 5, 2020. Pursuant to the terms of the A&R Brown Employment Agreement, the
Company agreed to pay Mr. Brown an annual salary of $300,000, less any applicable payroll deductions and tax withholdings (the “Brown
Base Salary”).
Either
the Company or Mr. Brown were permitted to terminate Mr. Brown’s employment with the Company at any time upon 30 days’ written notice
to the other party, subject to the terms of the A&R Brown Employment Agreement. In the event of such termination, the Company had
agreed to pay Mr. Brown the Brown Base Salary through the date of termination and other customary accrued and reimbursable expenses.
Additionally, in the event the Company terminated Mr. Brown’s employment for Cause (as defined in the A&R Brown Employment
Agreement) or the Mr. Brown voluntarily resigned for any reason on or before the six-month anniversary of the Brown Effective Date, the
Company would have had no further liability or obligation Mr. Brown under the A&R Brown Employment Agreement other than any customary
accrued and reimbursable expenses. In the event Mr. Brown’s employment was terminated by (x) the Company without Cause following
the Brown Effective Date or (y) Mr. Brown due to a resignation for any reason following the six-month anniversary of the Brown Effective
Date, subject to certain terms and conditions of the A&R Brown Employment Agreement, the Company agreed to pay Mr. Brown severance
pay in a total amount equal to $180,000.
Additionally,
pursuant to the A&R Brown Employment Agreement, Mr. Brown was not permitted to own over aggregate of 2% of the outstanding stock
of any class of any corporation engaged in a business that directly competes with the business of the Company if such stock is listed
on a national securities exchange in the United States.
The
A&R Brown Employment Agreement also provided for certain customary covenants regarding non-solicitation, non-competition and confidentiality.
For
the years ended December 31, 2025, and 2024, the Compensation Committee approved performance bonuses of $94,500 and $44,500, respectively
to Mr. Brown.
Retirement,
Health, Welfare and Additional Benefits
All
of our Named Executive Officers are eligible to participate in our employee benefit plans and programs, including medical benefits, to
the same extent as our other full-time employees, subject to the terms and eligibility requirements of those plans.
2004
Global Share Option Plan
In
November 2004, our Board adopted the 2004 Global Share Option Plan, pursuant to which 36,364 shares of our common stock were reserved
for issuance as awards to employees, directors, consultants and other service providers. The purpose of the 2004 Global Share Option
Plan was to provide an incentive to attract and retain directors, officers, consultants, advisors and employees, to encourage a sense
of proprietorship and stimulate an active interest of such persons in our development and financial success. The 2004 Global Share Option
Plan which was administered by our Board expired on February 28, 2014.
2014
Long-Term Incentive Plan
On
February 28, 2014, our stockholders approved the NanoVibronix, Inc. 2014 Long-Term Incentive Plan (the “2014 Plan”), which
was adopted by our Board on February 19, 2014.
Under
the 2014 Plan, we originally reserved a total of five million (5,000,000) shares of our common stock for issuance pursuant to awards
to key employees, key contractors, and non-employee directors, of which, the maximum number of shares of common stock covering awards
of stock options or stock appreciation rights that could be granted to certain of our executive officers during any calendar year was
one million (1,000,000) shares. On May 7, 2014, we effected a one-for-seven reverse stock split of our common stock. Consequently, the
number of shares of our common stock reserved for issuance pursuant to awards under the 2014 Plan was reduced to seven hundred fourteen
thousand two hundred eighty-six (714,286) shares, and the maximum number of shares of our common stock covered by awards of stock options
or stock appreciation rights that could be granted to certain of our executive officers during any calendar year was reduced to one hundred
forty-two thousand eight hundred fifty-seven (142,857) shares.
On
June 13, 2018, the stockholders approved an amendment to the 2014 Plan to increase the number of shares of our common stock reserved
for issuance pursuant to awards under the 2014 Plan by an additional seven hundred and fifty thousand (750,000) shares of our common
stock to one million four hundred sixty-four thousand two hundred eighty-six (1,464,286) shares.
On
June 13, 2019, the stockholders approved a second amendment to the 2014 Plan to increase (i) the number of shares of our common stock
available for issuance pursuant to awards under the 2014 Plan by four hundred thousand (400,000) shares of our common stock, to a total
of one million eight hundred and sixty-four thousand two hundred eighty-six (1,864,286) shares of our common stock and (ii) the maximum
number of shares of our common stock covering awards of stock options or stock appreciation rights that could be granted to certain of
our executive officers during any calendar year was increased to three hundred fifty-four thousand two hundred fourteen (354,214) shares.
On
December 29, 2021, the stockholders approved a third amendment to the 2014 Plan that (i) intended to increase the number of shares of
our common stock available for issuance pursuant to awards under the 2014 Plan by one million five hundred thousand (1,500,000) shares
of our common stock to a total of three million three hundred sixty-four thousand two-hundred eighty-six (3,364,286) shares of our common
stock, but a scrivener’s error in this amendment only increased the number of shares of our common stock available for issuance
pursuant to awards under the 2014 Plan to a total of three million three hundred forty-six thousand two-hundred eighty-six (3,346,286)
shares of our common stock, and (ii) increased the maximum number of shares of our common stock covering awards of stock options or stock
appreciation rights that could be granted to certain of our executive officers during any calendar year to six hundred sixty-nine thousand
two-hundred fifty-seven (669,257) shares of our common stock.
On
December 15, 2022, the stockholders approved a fourth amendment to the 2014 Plan to increase (i) the number of shares of our common stock
available for issuance pursuant to awards under the 2014 Plan by one million five hundred and eighteen thousand (1,518,000) shares of
our common stock, to a total of four million eight hundred and sixty-four thousand two hundred eighty-six (4,864,286) shares of our common
stock. On February 9, 2023, we effected a one-for-twenty reverse stock split of our common stock. Consequently, the number of shares
of our common stock reserved for issuance pursuant to awards under the 2014 Plan was reduced to two hundred forty-three thousand two
hundred fourteen (243,214) shares.
On
February 19, 2024, the 2014 Plan expired in accordance with its terms. Any awards granted on or before such date will continue to be
effective in accordance with their terms and conditions.
Description
of the 2014 Plan
Purpose.
The purpose of the 2014 Plan was to enable us to remain competitive and innovative in our ability to attract and retain the services
of key employees, key contractors, and non-employee directors. The 2014 Plan provided for the granting of incentive stock options, nonqualified
stock options, stock appreciation rights, restricted stock, restricted stock units, performance awards, dividend equivalent rights, and
other awards, which may be granted singly, in combination, or in tandem, and which may be paid in cash or shares of our common stock.
The 2014 Plan provided flexibility to our compensation methods in order to adapt the compensation of key employees, key contractors,
and non-employee directors to a changing business environment, after giving due consideration to competitive conditions and the impact
of applicable tax laws.
Effective
Date and Expiration. The 2014 Plan was originally approved by our Board on February 19, 2014, and became effective upon stockholder
approval on February 28, 2014. The 2014 Plan terminated on February 19, 2024. No award may be made under the 2014 Plan after its termination
date, but awards made prior to the termination date may extend beyond that date.
Share
Authorization. Subject to certain adjustments, the number of shares of our common stock that were reserved for issuance pursuant
to awards under the 2014 Plan was one million eight hundred and sixty-four thousand two hundred eighty- six (1,864,286) shares, of which
100% were able to be delivered pursuant to incentive stock options. Subject to certain adjustments, with respect to any participant who
is an officer of our company and subject to Section 16 of the Exchange Act, or a “covered employee” as defined in Section
162(m)(3) of the Internal Revenue Code of 1986, as amended (the “Code”), a maximum of three hundred fifty four thousand two
hundred fourteen (354,214) shares may be granted in any one year in the form of stock options or stock appreciation rights to such participant.
Shares
to be issued may be made available from authorized but unissued shares of our common stock, shares held by us in our treasury, or shares
purchased by us on the open market or otherwise. During the term of the 2014 Plan, we at all times reserved and kept enough shares available
to satisfy the requirements of the 2014 Plan. In the event that previously acquired shares were delivered to us in full or partial payment
of the option price for the exercise of a stock option granted under the 2014 Plan, the number of shares available for future awards
under the 2014 Plan would have been reduced only by the net number of shares issued upon the exercise of the stock option or settlement
of an award. Awards that may be satisfied either by the issuance of common stock or by cash or other consideration would have been counted
against the maximum number of shares that could have been issued under the 2014 Plan only during the period that the award is outstanding
or to the extent the award is ultimately satisfied by the issuance of shares.
Administration.
The 2014 Plan was administered by the compensation committee of our Board (the “Committee”). At any time there was no
Committee to administer the 2014 Plan, any reference to the Committee is a reference to the Board. The Committee would determine the
persons to whom awards are to be made; determine the type, size, and terms of awards; interpret the 2014 Plan; establish and revise rules
and regulations relating to the 2014 Plan and any sub-plans, including, without limitation, any sub-plans for awards made to participants
who are not residents of the United States; establish performance goals for awards and certify the extent of their achievement; and make
any other determinations that it believes necessary for the administration of the 2014 Plan. The Committee had the ability to delegate
certain duties to one or more of our officers as provided in the 2014 Plan.
Eligibility.
Employees (including any employee who is also a director or an officer), contractors, and non-employee directors of us or our subsidiaries
whose judgment, initiative, and efforts contributed to or may be expected to contribute to our successful performance were eligible to
participate in the 2014 Plan.
Stock
Options. The Committee had the ability to grant either incentive stock options (“ISOs”) qualifying under Section 422
of the Code or nonqualified stock options, provided that only employees of us and our subsidiaries (excluding subsidiaries that are not
corporations) are eligible to receive ISOs. Stock options could not be granted with an option price less than 100% of the fair market
value of a share of common stock on the date the stock option is granted. If an ISO was granted to an employee who owns or is deemed
to own more than 10% of the combined voting power of all classes of our stock (or any parent or subsidiary), the option price was to
be at least 110% of the fair market value of a share of common stock on the date of grant. The Committee had the ability to determine
the terms of each stock option at the time of grant, including, without limitation, the methods by or forms in which shares will be delivered
to participants. The maximum term of each option, the times at which each option will be exercisable, and provisions requiring forfeiture
of unexercised options at or following termination of employment or service generally were fixed by the Committee, except that the Committee
could not grant stock options with a term exceeding 10 years or, in the case of an ISO granted to an employee who owns or is deemed to
own more than 10% of the combined voting power of all classes of our stock (or any parent or subsidiary), a term exceeding five years.
Recipients
of stock options may pay the option price (i) in cash, check, bank draft, or money order payable to the order of the Company; (ii) by
delivering to us shares of Common Stock (included restricted stock) already owned by the participant having a fair market value equal
to the aggregate option price and that the participant has not acquired from us within six months prior to the exercise date; (iii) by
delivering to us or our designated agent an executed irrevocable option exercise form together with irrevocable instructions from the
participant to a broker or dealer, reasonably acceptable to us, to sell certain of the shares purchased upon the exercise of the option
or to pledge such shares to the broker as collateral for a loan from the broker and to deliver to us the amount of sale or loan proceeds
necessary to pay the purchase price; and (iv) by any other form of valid consideration that is acceptable to the Committee in its sole
discretion.
2024
Long-Term Incentive Plan
On
December 19, 2024, our stockholders approved the NanoVibronix, Inc. 2024 Long-Term Incentive Plan, which was adopted by our Board on
November 6, 2023, and on December 4, 2025, our stockholders approved the First Amendment (the “First Amendment”) to the 2024
Long-Term Incentive Plan (collectively, the “2024 Plan”).
Description
of the 2024 Plan
Purpose.
The purpose of the 2024 Plan is to enable us to remain competitive and innovative in our ability to attract and retain the services of
key employees, key contractors, and outside directors. The 2024 Plan provides for the granting of incentive stock options, nonqualified
stock options, stock appreciation rights, restricted stock, restricted stock units, performance awards, dividend equivalent rights, and
other awards, which may be granted singly, in combination, or in tandem, and which may be paid in cash or shares of our common stock.
The 2024 Plan is expected to provide flexibility to our compensation methods in order to adapt the compensation of key employees, key
contractors, and outside directors to a changing business environment, after giving due consideration to competitive conditions and the
impact of applicable tax laws.
Effective
Date and Expiration. The 2024 Plan was originally approved by our Board on November 6, 2023, subject to stockholder approval. The
2024 Plan will be effective upon approval by our stockholders (such date being, the “Effective Date”), and the 2024 Plan
will terminate on the tenth anniversary of the Effective Date, unless sooner terminated by our Board. No award may be made under the
2024 Plan after its termination date, but awards made prior to the termination date may extend beyond that date.
Share
Authorization. Subject to certain adjustments and to increase by any shares subject to Prior Plan Awards (defined below) that are
eligible for reuse, the number of shares of our common stock that are reserved for issuance pursuant to awards under the 2024 Plan is
1,205,454 shares, of which 100% may be delivered pursuant to incentive stock options. Prior to the First Amendment, the number of shares
of our common stock that were reserved for issuance pursuant to awards under the 2024 Plan was 600,000 shares. “Prior Plan Awards”
means (i) any awards under the Prior Plan (defined below) that are outstanding on the Effective Date and that, on or after the Effective
Date, are forfeited, expire, or are canceled; and (ii) any shares subject to awards relating to common stock under the Prior Plan that,
on or after the Effective Date, are settled in cash. “Prior Plan” means the 2014 Plan. Any awards outstanding under the Prior
Plan as of the Effective Date will continue to be governed by the terms and conditions of the Prior Plan and the applicable award agreement.
Shares
to be issued may be made available from authorized but unissued shares of our common stock, shares held by us in our treasury, or shares
purchased by us on the open market or otherwise. During the term of the 2024 Plan, we will at all times reserve and keep enough shares
available to satisfy the requirements of the 2024 Plan. If an award under the 2024 Plan or any Prior Plan Award is cancelled, forfeited,
or expires, in whole or in part, the shares subject to such forfeited, expired, or cancelled award may again be awarded under the 2024
Plan. In the event that previously acquired shares are delivered to us in full or partial payment of the option price for the exercise
of a stock option granted under the 2024 Plan, the number of shares available for future awards under the 2024 Plan shall be reduced
only by the net number of shares issued upon the exercise of the stock option or settlement of an award. Awards that may be satisfied
either by the issuance of common stock or by cash or other consideration shall be counted against the maximum number of shares that may
be issued under the 2024 Plan only during the period that the award is outstanding or to the extent the award is ultimately satisfied
by the issuance of shares. An award will not reduce the number of shares that may be issued pursuant to the 2024 Plan if the settlement
of the award will not require the issuance of shares, as, for example, a stock appreciation right that can be satisfied only by the payment
of cash. Only shares forfeited back to us; shares cancelled on account of termination, expiration, or lapse of an award; shares surrendered
in payment of the option price of an option; or shares withheld for payment of applicable employment taxes and/or withholding obligations
resulting from the exercise of a stock option shall again be available for grant as incentive stock options under the 2024 Plan, but
shall not increase the maximum number of shares described above as the maximum number of shares that may be delivered pursuant to incentive
stock options.
Limitation
on Outside Director Awards. Outside directors may not be granted awards under the 2024 Plan in any calendar year that exceed seven
hundred thousand dollars ($700,000) in the aggregate (with the fair market value of any equity awards determined as of the date of grant),
other than a one-time award granted to a newly appointed or elected outside director not to exceed an additional seven hundred thousand
dollars ($700,000) in the aggregate; provided, however, that these limits shall not apply to any awards made pursuant to a deferred compensation
arrangement in lieu of all or a portion of cash retainers otherwise payable to an outside director.
Administration.
The 2024 Plan is administered by the compensation committee of our Board (the “Committee”). At any time there is no Committee
to administer the 2024 Plan, any reference to the Committee is a reference to the Board. The Committee will determine the persons to
whom awards are to be made; determine the type, size, and terms of awards; interpret the 2024 Plan; establish and revise rules and regulations
relating to the 2024 Plan and any sub-plans, including, without limitation, any sub-plans for awards made to participants who are not
residents of the United States; establish performance goals for awards and certify the extent of their achievement; and make any other
determinations that it believes necessary for the administration of the 2024 Plan. The Committee may delegate certain duties to one or
more of our officers as provided in the 2024 Plan.
Eligibility.
Employees (including any employee who is also a director or an officer), contractors, and outside directors of us or our subsidiaries
whose judgment, initiative, and efforts contributed to or may be expected to contribute to our successful performance are eligible to
participate in the 2024 Plan. As of the date of this Annual Report on Form 10-K, we had 7 employees, 9 contractors, and 5 non-employee
directors who would be eligible for awards under the 2024 Plan.
Stock
Options. The Committee may grant either incentive stock options (“ISOs”) qualifying under Section 422 of the Internal
Revenue Code of 1986, as amended (the “Code”) or nonqualified stock options, provided that only employees of us and our subsidiaries
(excluding subsidiaries that are not corporations) are eligible to receive ISOs. Stock options may not be granted with an option price
less than 100% of the fair market value of a share of common stock on the date the stock option is granted. If an ISO is granted to an
employee who owns or is deemed to own more than 10% of the combined voting power of all classes of our stock (or any parent or subsidiary),
the option price shall be at least 110% of the fair market value of a share of common stock on the date of grant. The Committee will
determine the terms of each stock option at the time of grant, including, without limitation, the methods by or forms in which shares
will be delivered to participants. The maximum term of each option, the times at which each option will be exercisable, and provisions
requiring forfeiture of unexercised options at or following termination of employment or service generally are fixed by the Committee,
except that the Committee may not grant stock options with a term exceeding 10 years or, in the case of an ISO granted to an employee
who owns or is deemed to own more than 10% of the combined voting power of all classes of our stock (or any parent or subsidiary), a
term exceeding five years.
Recipients
of stock options may pay the option price (i) in cash, check, bank draft, or money order payable to the order of the Company; (ii) by
delivering to us shares of common stock (included restricted stock) already owned by the participant having a fair market value equal
to the aggregate option price and that the participant has not acquired from us within six months prior to the exercise date; (iii) by
delivering to us or our designated agent an executed irrevocable option exercise form together with irrevocable instructions from the
participant to a broker or dealer, reasonably acceptable to us, to sell certain of the shares purchased upon the exercise of the option
or to pledge such shares to the broker as collateral for a loan from the broker and to deliver to us the amount of sale or loan proceeds
necessary to pay the purchase price; and (iv) by any other form of valid consideration that is acceptable to the Committee in its sole
discretion.
Stock
Appreciation Rights. The Committee is authorized to grant stock appreciation rights (“SARs”) as a stand-alone award,
or freestanding SARs, or in conjunction with options granted under the 2024 Plan, or tandem SARs. SARs entitle a participant to receive
an amount equal to the excess of the fair market value of a share of common stock on the date of exercise over the fair market value
of a share of our common stock on the date of grant. The grant price of a SAR cannot be less than 100% of the fair market value of a
share of our common stock on the date of grant. The Committee will determine the terms of each SAR at the time of the grant, including,
without limitation, the methods by or forms in which shares will be delivered to participants. The maximum term of each SAR, the times
at which each SAR will be exercisable, and provisions requiring forfeiture of unexercised SARs at or following termination of employment
or service generally are fixed by the Committee, except that no freestanding SAR may have a term exceeding 10 years and no tandem SAR
may have a term exceeding the term of the option granted in conjunction with the tandem SAR. Distributions to the recipient may be made
in common stock, cash, or a combination of both as determined by the Committee.
Restricted
Stock and Restricted Stock Units. The Committee is authorized to grant restricted stock and restricted stock units. Restricted stock
consists of shares of our common stock that may not be sold, assigned, transferred, pledged, hypothecated, encumbered, or otherwise disposed
of, and that may be forfeited in the event of certain terminations of employment or service, prior to the end of a restricted period
as specified by the Committee. Restricted stock units are the right to receive shares of common stock at a future date in accordance
with the terms of such grant upon the attainment of certain conditions specified by the Committee, which include a substantial risk of
forfeiture and restrictions on their sale or other transfer by the participant. The Committee determines the eligible participants to
whom, and the time or times at which, grants of restricted stock or restricted stock units will be made; the number of shares or units
to be granted; the price to be paid, if any; the time or times within which the shares covered by such grants will be subject to forfeiture;
the time or times at which the restrictions will terminate; and all other terms and conditions of the grants. Restrictions or conditions
could include, but are not limited to, the attainment of performance goals (as described below), continuous service with us, the passage
of time, or other restrictions or conditions. Except as otherwise provided in the 2024 Plan or the applicable award agreement, a participant
shall have, with respect to shares of restricted stock, all of the rights of a stockholder of the Company holding the class of common
stock that is the subject of the restricted stock, including, if applicable, the right to vote the common stock and the right to receive
any dividends thereon.
Dividend
Equivalent Rights. The Committee is authorized to grant a dividend equivalent right to any participant, either as a component of
another award or as a separate award, conferring on the participant the right to receive credits based on the cash dividends that would
have been paid on the shares of common stock specified in the award as if such shares were held by the participant. The terms and conditions
of the dividend equivalent right shall be specified by the grant. Dividend equivalents credited to the holder of a dividend equivalent
right may be paid currently or may be deemed to be reinvested in additional shares. Any such reinvestment shall be at the fair market
value at the time thereof. A dividend equivalent right may be settled in cash, shares, or a combination thereof.
Performance
Awards. The Committee may grant performance awards payable in cash, shares of common stock, other consideration, or a combination
thereof at the end of a specified performance period. Payment will be contingent upon achieving pre-established performance goals (as
discussed below) by the end of the performance period. The Committee will determine the length of the performance period, the maximum
payment value of an award, and the minimum performance goals required before payment will be made, so long as such provisions are not
inconsistent with the terms of the 2024 Plan, and to the extent an award is subject to Section 409A of the Code, are in compliance with
the applicable requirements of Section 409A of the Code and any applicable regulations or guidance issued thereunder. In certain circumstances,
the Committee may, in its discretion, determine that the amount payable with respect to certain performance awards will be reduced from
the amount of any potential awards, if the Committee determines, in its sole discretion, that the established performance measures or
objectives are no longer suitable because of a change in our business, operations, corporate structure, or for other reasons that the
Committee deemed satisfactory, in which case, the Committee may modify the performance measures or objectives and/or the performance
period as the Committee deems appropriate in its sole discretion.
Performance
Goals. Awards under the 2024 Plan may be made subject to the attainment of performance goals relating to one or more business criteria
which, where applicable, shall consist of one or more or any combination of the following criteria (the “Performance Criteria”):
cash flow; cost; revenues; sales; ratio of debt to debt plus equity; net borrowing, credit quality, or debt ratings; profit before tax;
economic profit; earnings before interest and taxes; earnings before interest, taxes, depreciation, and amortization; gross margin; earnings
per share (whether on a pre-tax, after-tax, operational, or other basis); operating earnings; capital expenditures; expenses or expense
levels; economic value added; ratio of operating earnings to capital spending or any other operating ratios; free cash flow; net profit;
net sales; net asset value per share; the accomplishment of mergers, acquisitions, dispositions, public offerings, or similar extraordinary
business transactions; sales growth; price of the shares; return on assets, equity, or stockholders’ equity; market share; inventory
levels, inventory turn, or shrinkage; or total return to stockholders. Any Performance Criteria may be used to measure our performance
as a whole or any of our business units and may be measured relative to a peer group or index. Any Performance Criteria may include or
exclude (i) events that are of an unusual nature or indicate infrequency of occurrence; (ii) gains or losses on the disposition of a
business; (iii) changes in tax or accounting regulations or laws; (iv) the effect of a merger or acquisition, as identified in our quarterly
and annual earnings releases; or (v) other similar occurrences. In all
other respects, Performance Criteria shall be calculated in accordance with our financial statements, under generally accepted accounting
principles, or under a methodology established by the Committee prior to the issuance of an award, which is consistently applied and
identified in the Company’s audited financial statements, including in footnotes, or the Compensation Discussion and Analysis section
of the Company’s annual report.
Israeli
Awards
For
persons subject to the Israeli Income Tax Ordinance [New Version], 5721-1961 (the “Ordinance”), the Committee is authorized
to grant stock options pursuant to the terms of the Israeli Appendix. The Committee may grant to participants who are employees and office
holders options under Section 102 of the Ordinance (“Section 102 Options”) and to Controlling Shareholders (as defined in
the Israeli Appendix) and outside participants options under Section 3(i) of the Ordinance (“Section 3(i) Options”). The
Committee may designate Section 102 Options as “Approved 102 Options,” for which the options and shares upon exercise must
be held in trust and granted through a trustee, or as “Unapproved 102 Options,” for which the options and shares upon exercise
do not have to be held in trust. As described further below, the determination of the Committee as to the taxation route of the stock
options, the type of option, and duration of time the option and shares upon exercise are held in trust will determine the tax consequences
to the participant. Of the Approved 102 Options, the Committee may grant options as “Ordinary Income Options,” for which
the options and shares upon exercise must be held in trust for twelve (12) months from the date of grant, or as “Capital Gain Options,”
for which the options and shares upon exercise must be held in trust for twenty-four (24) months from the date of grant. If the requirements
of the Approved 102 Options are not met, the options are regarded as Unapproved 102 Options. Section 3(i) Options and the shares upon
exercise may, but need not, be held in trust as well, depending upon the agreement between the Committee, the participant, and the trustee
of the trust. Israeli participants can be granted other types of options under the 2024 Plan, but some of them will require a pre-ruling
from the Israeli Tax Authorities in order to be deemed Approved 102 Options.
Other
Awards. The Committee may grant other forms of awards, based upon, payable in, or that otherwise relate to, in whole or in part,
shares of common stock, if the Committee determines that such other form of award is consistent with the purpose and restrictions of
the 2024 Plan. The terms and conditions of such other form of award shall be specified by the grant. Such other awards may be granted
for no cash consideration, for such minimum consideration as may be required by applicable law, or for such other consideration as may
be specified by the grant.
Vesting,
Forfeiture, Recoupment and Assignment. The Committee, in its sole discretion, may determine that an award will be immediately vested
in whole or in part, or that all or any portion may not be vested until a date, or dates, subsequent to its date of grant, or until the
occurrence of one or more specified events, subject in any case to the terms of the 2024 Plan. If the Committee imposes conditions upon
vesting, then, except as otherwise provided below, subsequent to the date of grant, the Committee may, in its sole discretion, accelerate
the date on which all or any portion of the award may be vested.
The
Committee may impose on any award at the time of grant or thereafter, such additional terms and conditions as the Committee determines,
including terms requiring forfeiture of awards in the event of a participant’s termination of service. The Committee will specify
the circumstances on which performance awards may be forfeited in the event of a termination of service by a participant prior to the
end of a performance period or settlement of awards. Except as otherwise determined by the Committee, restricted stock will be forfeited
upon a participant’s termination of service during the applicable restriction period. In addition, we may recoup all or any portion
of any shares or cash paid to a participant in connection with any award in the event of a restatement of our financial statements as
set forth in our clawback policy, as such policy may be approved or modified by our Board from time to time.
Awards
granted under the 2024 Plan generally are not assignable or transferable except by will or by the laws of descent and distribution, except
that the Committee may, in its discretion and pursuant to the terms of an award agreement, permit transfers of certain award of nonqualified
stock options or SARs to (i) the spouse (or former spouse), children, or grandchildren of the participant (“Immediate Family Members”);
(ii) a trust or trusts for the exclusive benefit of such Immediate Family Members; (iii) a partnership in which the only partners are
(x) such Immediate Family Members, and/or (y) entities which are controlled
by Immediate Family Members; (iv) an entity exempt from federal income tax pursuant to Section 501(c)(3) of the Code or any successor
provision; or (v) a split interest trust or pooled income fund described in Section 2522(c)(2) of the Code or any successor provision,
provided that (x) there shall be no consideration for any such transfer, (y) the applicable award agreement pursuant to which such award
is granted must be approved by the Committee and must expressly provide for such transferability, and (z) subsequent transfers of transferred
awards shall be prohibited except those by will or the laws of descent and distribution.
Adjustments
Upon Changes in Capitalization. In the event that any dividend or other distribution, recapitalization, stock split, reverse stock
split, rights offering, reorganization, merger, consolidation, split-up, spin-off, split-off, combination, subdivision, repurchase, or
exchange of shares of common stock or other securities of the Company, issuance of warrants or other rights to purchase shares of common
stock or other securities of the Company, or other similar corporate transaction or event affects the fair value of an award, then the
Committee shall adjust any or all of the following so that the fair value of the award immediately after the transaction or event is
equal to the fair value of the award immediately prior to the transaction or event (i) the number of shares and type of common stock
(or the securities or property) which thereafter may be made the subject of awards; (ii) the number of shares and type of common stock
(or other securities or property) subject to outstanding awards; (iii) the number of shares and type of common stock (or other securities
or property) specified as the annual per-participant limitation under the 2024 Plan; (iv) the option price of each outstanding award;
(v) the amount, if any, we pay for forfeited shares in accordance with the terms of the 2024 Plan; and (vi) the number of or exercise
price of shares then subject to outstanding SARs previously granted and unexercised under the 2024 Plan to the end that the same proportion
of our issued and outstanding shares of common stock in each instance shall remain subject to exercise at the same aggregate exercise
price; provided, however, that the number of shares of common stock (or other securities or property) subject to any award shall always
be a whole number. Notwithstanding the foregoing, no such adjustment shall be made or authorized to the extent that such adjustment would
cause the 2024 Plan or any stock option to violate Section 422 of the Code or Section 409A of the Code. All such adjustments must be
made in accordance with the rules of any securities exchange, stock market, or stock quotation system to which we are subject.
Amendment
or Discontinuance of the 2024 Plan. The Board may, at any time and from time to time, without the consent of participants, alter,
amend, revise, suspend, or discontinue the 2024 Plan in whole or in part; provided, however, that (i) no amendment that requires stockholder
approval in order for the 2024 Plan and any awards under the 2024 Plan to continue to comply with Sections 421 and 422 of the Code (including
any successors to such sections, or other applicable law) or any applicable requirements of any securities exchange or inter-dealer quotation
system on which our stock is listed or traded, shall be effective unless such amendment is approved by the requisite vote of our stockholders
entitled to vote on the amendment; and (ii) unless required by law, no action by the Board regarding amendment or discontinuance of the
2024 Plan may adversely affect any rights of any participants or obligations of us to any participants with respect to any outstanding
awards under the 2024 Plan without the consent of the affected participant.
U.S.
Federal Income Tax Consequences
The
following is a brief summary of certain U.S. federal income tax consequences relating to the transactions described under the 2024 Plan
as set forth below. This summary does not purport to address all aspects of U.S. federal income taxation and does not describe state,
local, or foreign tax consequences. This discussion is based upon provisions of the Code and the Treasury Regulations issued thereunder,
and judicial and administrative interpretations under the Code and Treasury Regulations, all as in effect as of the date hereof, and
all of which are subject to change (possibly on a retroactive basis) or different interpretation.
Law
Affecting Deferred Compensation. In 2004, Section 409A was added to the Code to regulate all types of deferred compensation. If the
requirements of Section 409A of the Code are not satisfied, deferred compensation and earnings thereon will be subject to tax as it vests,
plus an interest charge at the underpayment rate plus 1% and a 20% penalty tax. Certain performance awards, stock options, stock appreciation
rights, restricted stock units, and certain types of restricted stock are subject to Section 409A of the Code.
Incentive
Stock Options. A participant will not recognize income at the time an ISO is granted. When a participant exercises an ISO, a participant
also generally will not be required to recognize income (either as ordinary income or capital gain). However, to the extent that the
fair market value (determined as of the date of grant) of the shares with respect to which the participant’s ISOs are exercisable
for the first time during any year exceeds $100,000, the ISOs for the shares over $100,000 will be treated as nonqualified stock options,
and not ISOs, for U.S. federal tax purposes, and the participant will recognize income as if the ISOs were nonqualified stock options.
In addition to the foregoing, if the fair market value of the shares received upon exercise of an ISO exceeds the exercise price, then
the excess may be deemed a tax preference adjustment for purposes of the U.S. federal alternative minimum tax calculation. The federal
alternative minimum tax may produce significant tax repercussions depending upon the participant’s particular tax status.
The
tax treatment of any shares acquired by exercise of an ISO will depend upon whether the participant disposes of his or her shares prior
to the later of: (i) two years after the date the ISO was granted or (ii) one year after the shares were transferred to the participant
upon exercise of the ISO (referred to as the “Holding Period”). If a participant disposes of shares acquired by exercise
of an ISO after the expiration of the Holding Period, any amount received in excess of the participant’s tax basis for such shares
will be treated as a short-term or long-term capital gain, depending upon how long the participant has held the shares. If the amount
received is less than the participant’s tax basis for such shares, the loss will be treated as a short-term or long-term capital
loss, depending upon how long the participant has held the shares. If the participant disposes of shares acquired by exercise of an ISO
prior to the expiration of the Holding Period, the disposition will be considered a “disqualifying disposition.” If the amount
received for the shares is greater than the fair market value of the shares on the exercise date, then the difference between the ISO’s
exercise price and the fair market value of the shares at the time of exercise will be treated as ordinary income for the tax year in
which the “disqualifying disposition” occurs. The participant’s basis in the shares will be increased by an amount
equal to the amount treated as ordinary income due to such “disqualifying disposition.” In addition, the amount received
in such “disqualifying disposition” over the participant’s increased basis in the shares will be treated as capital
gain. However, if the price received for shares acquired by exercise of an ISO is less than the fair market value of the shares on the
exercise date and the disposition is a transaction in which the participant sustains a loss which otherwise would be recognizable under
the Code, then the amount of ordinary income that the participant will recognize is the excess, if any, of the amount realized on the
“disqualifying disposition” over the basis of the shares.
Nonqualified
Stock Options. A participant generally will not recognize income at the time a nonqualified stock option is granted. When a participant
exercises a nonqualified stock option, the difference between the option price and any higher market value of the shares of common stock
on the date of exercise will be treated as compensation taxable as ordinary income to the participant. The participant’s tax basis
for the shares acquired under a nonqualified stock option will be equal to the option price paid for such shares, plus any amounts included
in the participant’s income as compensation. When a participant disposes of shares acquired by exercise of a nonqualified stock
option, any amount received in excess of the participant’s tax basis for such shares will be treated as short-term or long-term
capital gain, depending upon how long the participant has held the shares. If the amount received is less than the participant’s
tax basis for such shares, the loss will be treated as a short-term or long-term capital loss, depending upon how long the participant
has held the shares.
Special
Rule if Option Price is Paid for in Shares. If a participant pays the option price of a nonqualified stock option with previously-owned
shares of our common stock and the transaction is not a disqualifying disposition of shares previously acquired under an ISO, the shares
received equal to the number of shares surrendered are treated as having been received in a tax-free exchange. The participant’s
tax basis and holding period for these shares received will be equal to the participant’s tax basis and holding period for the
shares surrendered. The shares received in excess of the number of shares surrendered will be treated as compensation taxable as ordinary
income to the participant to the extent of their fair market value. The participant’s tax basis in these shares will be equal to
their fair market value on the date of exercise, and the participant’s holding period for such shares will begin on the date of
exercise.
If
the use of previously acquired shares to pay the exercise price of a nonqualified stock option constitutes a disqualifying disposition
of shares previously acquired under an ISO, the participant will have ordinary income as a result of the disqualifying disposition in
an amount equal to the excess of the fair market value of the shares surrendered, determined at the time such shares were originally
acquired on exercise of the ISO, over the aggregate option price paid for such shares. As discussed above, a disqualifying disposition
of shares previously acquired under an ISO occurs when the participant disposes of such shares before the end of the Holding Period.
The other tax results from paying the exercise price with previously-owned shares are as described above, except that the participant’s
tax basis in the shares that are treated as having been received in a tax-free exchange will be increased by the amount of ordinary income
recognized by the participant as a result of the disqualifying disposition.
Restricted
Stock. A participant who receives restricted stock generally will recognize as ordinary income the excess, if any, of the fair market
value of the shares granted as restricted stock at such time as the shares are no longer subject to forfeiture or restrictions, over
the amount paid, if any, by the participant for such shares. However, a participant who receives restricted stock may make an election
under Section 83(b) of the Code within 30 days of the date of transfer of the shares to recognize ordinary income on the date of transfer
of the shares equal to the excess of the fair market value of such shares (determined without regard to the restrictions on such shares)
over the purchase price, if any, of such shares. If a participant does not make an election under Section 83(b) of the Code, then the
participant will recognize as ordinary income any dividends received with respect to such shares. At the time of sale of such shares,
any gain or loss realized by the participant will be treated as either short-term or long-term capital gain (or loss) depending on the
holding period. For purposes of determining any gain or loss realized, the participant’s tax basis will be the amount previously
taxable as ordinary income, plus the purchase price paid by the participant, if any, for such shares.
Stock
Appreciation Rights. Generally, a participant who receives a stand-alone SAR will not recognize taxable income at the time the stand-alone
SAR is granted, provided that the SAR is exempt from or complies with Section 409A of the Code. If an employee receives the appreciation
inherent in the SARs in cash, the cash will be taxed as ordinary income to the recipient at the time it is received. If a recipient receives
the appreciation inherent in the SARs in stock, the spread between the then current market value and the grant price, if any, will be
taxed as ordinary income to the employee at the time it is received. In general, there will be no federal income tax deduction allowed
to us upon the grant or termination of SARs. However, upon the exercise of a SAR, we will be entitled to a deduction equal to the amount
of ordinary income the recipient is required to recognize as a result of the exercise.
Other
Awards. In the case of an award of restricted stock units, performance awards, dividend equivalent rights, or other stock or cash
awards, the recipient will generally recognize ordinary income in an amount equal to any cash received and the fair market value of any
shares received on the date of payment or delivery, provided that the award is exempt from or complies with Section 409A of the Code.
In that taxable year, we will receive a federal income tax deduction in an amount equal to the ordinary income which the participant
has recognized.
Federal
Tax Withholding. Any ordinary income realized by a participant upon the exercise of an award under the 2024 Plan is subject to withholding
of U.S. federal, state, and local income tax and to withholding of the participant’s share of tax under the Federal Insurance Contribution
Act and the Federal Unemployment Tax Act. To satisfy our federal income tax withholding requirements, we will have the right to require
that, as a condition to delivery of any certificate for shares of common stock or the registration of the shares in the participant’s
name, the participant remit to us an amount sufficient to satisfy the withholding requirements. Alternatively, we may withhold a portion
of the shares (valued at fair market value) that otherwise would be issued to the participant to satisfy all or part of the withholding
tax obligations or may, if we consent, accept delivery of shares (that the participant has not acquired from us within six months prior
to the date of exercise) with an aggregate fair market value that equals or exceeds the required tax withholding payment. Withholding
does not represent an increase in the participant’s total income tax obligation, since it is fully credited toward his or her tax
liability for the year. Additionally, withholding does not affect the participant’s tax basis in the shares. Compensation income
realized and tax withheld will be reflected on Forms W-2 supplied by us to employees by January 31 of the succeeding year. Deferred compensation
that is subject to Section 409A of the Code will be subject to certain federal income tax withholding and reporting requirements.
Tax
Consequences to Us. To the extent that a participant recognizes ordinary income in the circumstances described above, we will be entitled
to a corresponding deduction provided that, among other things, the income meets the test of reasonableness, is an ordinary and necessary
business expense, is not an “excess parachute payment” within the meaning of Section 280G of the Code, and is not disallowed
by the $1,000,000 limitation on certain executive compensation under Section 162(m) of the Code. While deductibility of executive compensation
for federal income tax purposes is among the factors the Board and Committee consider when structuring executive compensation arrangements,
it is not the sole or primary factor considered. We retain the flexibility to authorize compensation that may not be deductible if we
believe it is in the best interests of the Company.
Million
Dollar Deduction Limit and Other Tax Matters. We may not deduct compensation of more than $1,000,000 that is paid to “covered employees”
(as defined in Section 162(m) of the Code), which include (i) an individual (or, in certain circumstances, his or her beneficiaries)
who, at any time during the taxable year, is either our principal executive officer or principal financial officer; (ii) an individual
who is among our three highest compensated officers for the taxable year (other than an individual who was either our principal executive
officer or principal financial officer at any time during the taxable year); or (iii) anyone who was a covered employee for purposes
of Section 162(m) of the Code for any tax year beginning on or after January 1, 2018. This limitation on deductions (x) only applies
to compensation paid by a publicly-traded corporation (and not compensation paid by non-corporate entities) and (z) may not apply to
certain types of compensation, such as qualified performance-based compensation that is payable pursuant to a written, binding contract
that was in effect as of November 2, 2018, so long as the contract is not materially modified after that date.
If
an individual’s rights under the 2024 Plan are accelerated as a result of a change in control and the individual is a “disqualified
individual” under Section 280G of the Code, the value of any such accelerated rights received by such individual may be included
in determining whether or not such individual has received an “excess parachute payment” under Section 280G of the Code,
which could result in (i) the imposition of a 20% federal excise tax (in addition to federal income tax) payable by the individual on
the value of such accelerated rights, and (ii) the loss by us of a compensation deduction.
Israeli
Income Tax Consequences
The
following description of the Israel income tax consequences of awards under Israeli Appendix of the 2024 Plan is general and does not
purport to be complete.
Pursuant
to Section 102 of the Ordinance, which came into effect on January 1, 2003, options, shares, and other securities (including Restricted
Shares) (together “Options”) may be granted through a trustee (i.e., Approved 102 Options) or not through a trustee (i.e.,
Unapproved 102 Options). The following is a brief discussion of the tax consequences applicable to both types of Section 102 Options.
Grant
Through a Trustee
Options
granted through a trustee and held in trust are made either through the capital gains tax track (i.e., Capital Gains Options) or the
compensation income tax track (i.e., Ordinary Income Options). Capital Gains Options and Ordinary Income Options can be granted only
through a trustee. Under the capital gains tax track, the Capital Gains Options and the underlying shares have to be held in trust for
at least twenty-four (24) months from their date of grant. Any gain made on the sale of shares following the twenty-four (24) month period
is subject to a capital gains tax at a current rate of 25%; the amount of gain is the difference between the sales proceeds from the
sale of shares and the exercise price paid for such shares. Generally, Capital Gains Options are not taxed on their date of grant. However,
in the event that the exercise price of the options is less than the fair market value of the Company’s common stock on the date
of grant, a portion of the gain will be deemed compensation income, taxable at the personal marginal tax rate of the participant. The
payment of such tax is made at the time of exercise of the Capital Gains Options. The portion of the gain that is deemed compensation
income is the difference between the average value of the shares as listed on the stock exchange during the thirty (30) day period prior
to the date of grant and the exercise price of the option. If the Capital
Gains Options or the underlying shares of such options are sold by the trustee or transferred from the trustee to the beneficiary before
the end of the twenty-four (24) month period, any resulting income (cash or equivalent) is taxed as compensation income. If the options
have not been exercised and transferred from the trustee to the beneficiary, the taxable amount of income is the value of the option.
If the options have been exercised, the taxable amount of income is the difference between the aggregate fair market value of the shares
at the time of such sale or transfer and the aggregate exercise price paid for such shares.
Under
the compensation income tax track, the Ordinary Income Options and the underlying shares have to be held in trust for at least twelve
(12) months from their date of grant. Any gain made on the sale of shares is subject to compensation income tax at the personal marginal
tax rate of the respective participant; the amount of gain is the difference between the sales proceeds from the sale of shares and the
exercise price paid for such shares. Ordinary Income Options are not taxed on their date of grant, but rather when the options or the
underlying shares of such options are sold by the trustee or transferred from the trustee to the beneficiary. At such time, if the options
have not been exercised, the taxable amount of income is value of the Option. If the Options have been exercised, the taxable amount
of income is the difference between the aggregate fair market value of the shares at the time of such sale or transfer and the aggregate
exercise price paid for such shares.
A
corporate tax deduction is available for the employer in the tax year in which tax is withheld. The deductible amount is equal to any
amount included by a participant as compensation income, except when a participant is granted Capital Gains Options, including in the
event that such Capital Gains Options or the underlying shares of such options are sold by the trustee or transferred from the trustee
to the beneficiary before the end of the applicable twenty-four (24) month period. In such event, any resulting income to the participant
is deemed to be compensation income for tax purposes, but there would be no corresponding corporate tax deduction available to the employer.
Grant
Not Through a Trustee
In
the case of Options not made through a trustee, if the shares are non-marketable securities, the Option will not be subject to tax at
the date of grant of the option or the exercise of the Option. However, ordinary income tax will be payable upon the sale of the shares
acquired upon exercise of the Option. The taxable amount will be the sales proceeds less the aggregate exercise price paid by the participant.
If the shares covered by the option have a market value, then the value of the Option is treated as compensation income, and subject
to tax at the date of grant. There is no tax upon the exercise of the Option. However, capital gains tax will be payable on the sale
of the shares upon exercise of the Option. The taxable amount will be the sales proceeds, less the value that was taxed at the date of
grant and the aggregate exercise price paid by the participant.
Grant
of Section 3(i) Options
Options
under Section 3(i) of the Ordinance may be granted to Controlling Shareholders, consultants, and controlling stockholders (which are
excluded from the term employees under Section 102 of the Ordinance). Grants of Options for shares which are non-marketable are not taxed
under the income tax rules on the date of grant, but such event creates VAT liability. However, they are subject to tax at the time of
exercise at the ordinary income tax rate, and at the day such shares are sold at the capital gains tax rate. The difference between the
fair market value of the shares at the time of exercise and the exercise price is taxed at the ordinary income tax rate. Any gain above
such value at the time of sale of the shares is taxed at the capital gains rate. Grants of Options for shares which have a market value
are subject to tax on the date of grant, exercise of the Option, and the sale of the shares. The value of the Option is taxed on the
date of grant at the ordinary income tax rate. The difference between the fair market value of the shares at the time of exercise and
the sum of the exercise price and the amounts previously taxed at grant, is taxed at the ordinary income tax rate. Any gain above such
value at the time of sale of the shares is taxed at the capital gains rate.
Other
Stock Incentives
All
other awards under the Israeli Appendix need tax ruling from the Israeli Tax Authority for the postponement of the tax event arising
from the issuance thereof. Otherwise, there is an immediate tax event.
Outstanding
Equity Awards at December 31, 2025
The
following table provides certain information as of December 31, 2025, with respect to our equity compensation plans under which our equity
securities were authorized for issuance:
| | |
(a) | | |
(b) | | |
(c) | |
| Plan
Category | |
Number
of securities to be
issued upon exercise of outstanding options,
warrants, and rights | | |
Weighted-average
exercise price of outstanding options, warrants and rights | | |
Number
of securities remaining available
for future issuance under equity compensation plans (excluding securities reflected in column
(a)) | |
| Equity
compensation plans approved by security holders(1) | |
| 4,411 | | |
$ | 8.43 | | |
| 1,044 | |
| Equity
compensation plans not approved by security holders | |
| - | | |
| - | | |
| - | |
| Total | |
| 4,411 | | |
$ | 8.43 | | |
| 1,044 | |
| (1) |
Represents
shares available for issuance under the 2014 Plan and the 2024 Plan as of December 31, 2025, pursuant to outstanding awards. The
2014 Plan expired on February 19, 2024, and no further equity awards may be granted under the 2014 Plan. |
DIRECTOR
COMPENSATION
The
following table shows the compensation earned by persons who served on our Board during the fiscal year ended December 31, 2025, who
are not one of our Named Executive Officers. Other than as set forth in the table and described more fully below, we did not pay any
compensation, reimburse any expense of, make any equity awards or non-equity awards to, or pay any other compensation to any of the other
members of our Board for their services rendered in such period.
| | |
Fees
earned or | | |
Option
Awards | | |
| |
| Name | |
paid in cash ($) | | |
($) (1) | | |
Total ($) | |
| | |
| | | |
| | | |
| | |
| Current
Directors | |
| | | |
| | | |
| | |
| David
Johnson | |
$ | - | | |
$ | - | | |
$ | - | |
| | |
| | | |
| | | |
| | |
| Zeev
Rotstein, M.D. | |
$ | - | | |
$ | - | | |
$ | - | |
| | |
| | | |
| | | |
| | |
| Allison
Burgett | |
$ | - | | |
$ | - | | |
$ | - | |
| | |
| | | |
| | | |
| | |
Nino
Pionati | |
$ | - | | |
$ | - | | |
$ | - | |
| | |
| | | |
| | | |
| | |
| Former
Directors | |
| | | |
| | | |
| | |
| Christopher
Fashek (2) | |
$ | 260,500 | | |
$ | - | | |
$ | 260,500 | |
| | |
| | | |
| | | |
| | |
| Thomas
Mika (3) | |
$ | 95,000 | | |
$ | - | | |
$ | 95,000 | |
| | |
| | | |
| | | |
| | |
| Michael
Ferguson (4) | |
$ | 20,000 | | |
$ | - | | |
$ | 20,000 | |
| | |
| | | |
| | | |
| | |
| Martin
Goldstein (5) | |
$ | 60,000 | | |
$ | - | | |
$ | 60,000 | |
| | |
| | | |
| | | |
| | |
| Harold
Jacob, M. D. (6) | |
$ | 10,000 | | |
$ | - | | |
$ | 10,000 | |
| | |
| | | |
| | | |
| | |
| Aurora
Cassirer (7) | |
$ | 5,000 | | |
$ | - | | |
$ | 5,000 | |
| | |
| | | |
| | | |
| | |
| Maria
Schroeder (8) | |
$ | 10,000 | | |
$ | - | | |
$ | 10,000 | |
(1) In
accordance with SEC rules, the amounts in this column reflect the dollar amounts to be recognized for financial statement reporting
purposes with respect to the 2023 fiscal year in accordance with ASC Topic 718. Fair value is based on the Black-Scholes option
pricing model using the market price of the underlying shares at the grant date. For additional discussion of the valuation
assumptions used in determining stock-based compensation and the grant date fair value for stock options, see
“Management’s Discussion and Analysis of Financial Condition and Results of Operation - Critical Accounting Policies
- Stock-based compensation” and Note 3- “Summary of Significant Accounting Policies” and Note 11-
“Stockholders’ Equity” to our audited consolidated financial statements for the fiscal year ended December
31, 2024.
(2)
As of December 31, 2025, Mr. Fashek had outstanding options representing the right to purchase 582 shares of our common stock
and no outstanding stock awards of shares of common stock. Includes termination costs of $100,000.
(3)
As of December 31, 2025, Mr. Mika had outstanding options representing the right to purchase 364 shares of our common stock and
no outstanding stock awards of shares of common stock. Includes termination costs of $75,000.
(4)
As of December 31, 2025, Mr. Ferguson had outstanding options representing the right to purchase 364 shares of our common stock
and no outstanding stock awards of shares of common stock.
(5)
As of December 31, 2025, Mr. Goldstein had outstanding options representing the right to purchase 391 shares of our common stock
and no outstanding stock awards of shares of common stock. Includes termination costs of $50,000.
(6)
As of December 31, 2025, Dr. Jacob had outstanding options representing the right to purchase 182 shares of our common stock
and no outstanding stock awards of shares of common stock.
(7)
As of December 31, 2025, Ms. Cassirer had outstanding options representing the right to purchase 364 shares of our common stock
and no outstanding stock awards of shares of common stock.
(8)
As of December 31, 2025, Ms. Schroeder had outstanding options representing the right to purchase 364 shares of our common stock
and no outstanding stock awards of shares of common stock.
On
October 13, 2016, we entered into an agreement with Christopher Fashek to serve as the chairman of our Board. Under this agreement Mr.
Fashek was paid $100,000 per year payable in semi-monthly installments. On November 1, 2018, the Compensation committee voted to increase
Mr. Fashek’s consulting fee to $150,000 per year.
On
November 29, 2023, we entered into an option cancellation and release agreement with each of Brian Murphy, Christopher Fashek, Martin
Goldstein, Michael Ferguson, Stephen Brown, Aurora Cassirer, Dr. Harold Jacob, Maria Schroeder and Thomas Mika (collectively, the “Option
Holders”), pursuant to which the parties agreed to cancel options to purchase an aggregate of 9,276 shares of common stock at exercise
prices ranging from $98.34 to $565.40 (the “Options”) previously granted to each of the Option Holders. In exchange for the
cancellation of the Options, we paid $1.00 to each Option Holder.
No
other compensation was paid to our non-employee directors other than as noted in the table above for the one-year period ended December
31, 2025.
ITEM
12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS.
The
following table sets forth information with respect to the beneficial ownership of our common stock as of March 31, 2026, by:
| ● |
each
person known by us to beneficially own more than 5.0% of our common stock; |
| |
|
| ● |
each
of our directors; |
| |
|
| ● |
each
of our Named Executive Officers; and |
| |
|
| ● |
all
of our directors and executive officers as a group. |
The
percentages of common stock beneficially owned are reported on the basis of regulations of the SEC governing the determination of beneficial
ownership of securities. Under the rules of the SEC, a person is deemed to be a beneficial owner of a security if that person has or
shares voting power, which includes the power to vote or to direct the voting of the security, or investment power, which includes the
power to dispose of or to direct the disposition of the security.
Except
as indicated in the footnotes to this table, each beneficial owner named in the table below has sole voting and sole investment power
with respect to all shares beneficially owned and each person’s address is c/o ENvue Medical, Inc., 969 Pruitt Place, Tyler TX 75703.
As of March 31, 2026, we had 3,700,908 shares of common stock, 0 shares of Series C Preferred Stock, 0 shares of Series D Preferred Stock,
0 shares of Series E Preferred Stock, 0 shares of Series F Preferred Stock, 820 shares of Series G Preferred Stock, 10,209 shares of
Series H Preferred Stock and 50,528 shares of Series X Preferred Stock outstanding.
| Name
of Beneficial Owner | |
Number
of Shares Beneficially Owned (1) | | |
Percentage
of Shares Outstanding (1) | |
| 5% Owners | |
| | | |
| | |
| Alpha Capital Anstalt | |
| 181,344 | (2) | |
| 4.9 | %(2) |
| Directors and Executive
Officers | |
| | | |
| | |
| Doron Besser, M.D. | |
| 92,964 | (3) | |
| 2.5 | % |
| Nicole Fernandez-McGovern | |
| - | | |
| - | |
| Rita Silberberg | |
| - | | |
| - | |
| David Johnson | |
| - | | |
| - | |
| Alison Burgett | |
| - | | |
| - | |
| Nino Pionati | |
| - | | |
| - | |
| Zeev Rotstein, M.D. | |
| - | | |
| - | |
| Brian Murphy | |
| 909 | (4) | |
| * | % |
| Stephen Brown | |
| 582 | (5) | |
| * | % |
| All
directors and executive officers as a group (9 persons) | |
| 94,455 | | |
| 2.6 | % |
| * |
Represents
ownership of less than 1% |
| |
|
| (1) |
Shares of common stock beneficially owned and the respective percentages of beneficial ownership of common stock assume the exercise of
all options, warrants and other securities convertible into common stock beneficially owned by such person or entity currently exercisable
or exercisable within 60 days of March 31, 2026. Shares issuable pursuant to the exercise of stock options and warrants exercisable within
60 days are deemed outstanding and held by the holder of such options or warrants for computing the percentage of outstanding common stock
beneficially owned by such person, but are not deemed outstanding for computing the percentage of outstanding common stock beneficially
owned by any other person. |
| (2) |
The shares are held by Alpha Capital Anstalt, a company based in Vaduz Liechtenstein,
comprised of 0 shares of common stock, 39,002 Series X Preferred Shares, 10,209 Series H Preferred Shares and 181,344 shares of common
stock issuable upon the exercise of the February and July 2025 Warrants and excludes 570,020 shares of common stock issuable upon the
exercise of the 2025 Warrants which are subject to the beneficial ownership limitation of 4.9% as well as shares of common stock issuable
upon the exercise of the Preferred Stock The address of Alpha Capital Anstalt is Lettstrasse 32, FL-9490, Furstentums, Vaduz, Austria,
Liechtenstein. |
| |
|
| (3) |
Comprised of 3,128 Series X Preferred Shares to be converted into common stock held by Dr. Besser as of March 31,
2026. |
| |
|
| (4) |
Comprised of 582 shares of common stock that may be purchased by Mr. Brown
upon exercise of stock options that are currently exercisable or exercisable within 60 days following March 31, 2026. |
| |
|
| (5) |
Comprised of 909 shares of common stock that may be purchased by Mr. Murphy
upon exercise of stock options that are currently exercisable or exercisable within 60 days following March 31, 2026. |
ITEM
13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE.
Related
Parties Transactions Approval Policy
Generally,
we do not enter into related party transactions unless the members of the Board who do not have an interest in the potential transaction
have reviewed the transaction and determined that (i) we would not be able to obtain better terms by engaging in a transaction with a
non-related party and (ii) the transaction is in our best interest. In approving or rejecting any such proposal, our Board considers
all of the relevant facts and circumstances of the related party transaction and the related party’s relationship and interest
in the transaction. This policy applies generally to any transaction in which we are to be a participant and the amount involved exceeds
the lesser of $120,000 or one percent of the average of our total assets at year-end for the previous two completed fiscal years, and
in which any related person had or will have a direct or indirect material interest. This policy is not currently in writing.
The
Audit Committee is charged with reviewing, approving and overseeing any transaction between us and any related person (as defined in
Item 404 of Regulation S-K) and any other potential conflict of interest situations in accordance with Company policies and procedures.
All of the transactions described below were entered into prior to the establishment of our audit committee and were evaluated in accordance
with the policy described in the paragraph above. Prior to approving such transactions, the material facts as to a director’s or
officer’s relationship or interest as to the agreement or transaction were disclosed to our Board. Our Board took this information
into account when evaluating the transaction and in determining whether such transaction was fair to us and in the best interest of all
of our stockholders.
Transactions
with Related Parties
In
2025, we engaged the law firm Pierson Ferdinand to handle certain of our legal services. As has been previously disclosed, one of our
former board members, Aurora Cassirer, is a partner at Pierson Ferdinand. Ms. Cassirer did not provide any legal services or legal advice
to the Company. For the year ended December 31, 2025, we paid legal fees to Pierson Ferdinand in the amount of $323,000.
Other
than compensation agreements and other arrangements which are described as required under “Director Compensation” and “Executive
Compensation” and the transactions described above, since January 1, 2024, there has not been, and there is not currently proposed,
any transaction or series of similar transactions to which we were or will be a party in which the amount involved exceeded or will exceed
the lesser of $120,000 or the average of our total assets at year-end for the last two completed fiscal years and in which any director,
executive officer, holder of 5% or more of any class of our capital stock, or any member of their immediate family had or will have a
direct or indirect material interest.
ITEM
14. PRINCIPAL ACCOUNTING FEES AND SERVICES.
On
August 12, 2025, the Audit Committee of the Board of Directors approved the dismissal of Zwick CPA PLLC (“Zwick”) as the
Company’s independent registered public accounting firm, effective as of the same date.
The
reports of Zwick on the Company’s consolidated financial statements for the two most recent fiscal years, ended December 31, 2024,
and December 31, 2023, did not contain an adverse opinion or a disclaimer of opinion, and was not qualified or modified as to uncertainty,
audit scope, or accounting principles, except Zwick’s report on the consolidated financial statements of the Company as of and
for the years ended December 31, 2024, and December 31, 2023, contained an explanatory paragraph stating there was substantial doubt
about the Company’s ability to continue as a going concern.
During
the two most recent fiscal years, ended December 31, 2024, and December 31, 2023, and the subsequent interim period through August 12,
2025, there were no disagreements (as defined in Item 304(a)(1)(iv) of Regulation S-K and the related instructions to Item 304 of Regulation
S-K) with Zwick on any matter of accounting principles or practices, financial statement disclosure, or auditing scope or procedure,
which disagreements, if not resolved to the satisfaction of Zwick, would have caused Zwick to make reference to the subject matter of
the disagreements in connection with its reports on the Company’s consolidated financial statements for such years. Also during
this time, there were no “reportable events,” as defined in Item 304(a)(1)(v) of Regulation S-K, except that, for the years
ended December 31, 2024, and December 31, 2023, and for each of the quarters within the years ended December 31, 2024, and 2023, management
identified deficiencies in the Company’s design and effectiveness of their internal control over financial reporting that were
considered to be material weaknesses.
On
August 13, 2025, the Audit Committee engaged Kost Forer Gabbay & Kasierer,
a member of Ernst & Young Global (“EY”), as the Company’s independent registered public accounting firm for the
fiscal year ending December 31, 2025, effective immediately. EY also served as the Company’s independent registered public accounting
firm for the re-audit of the fiscal year ended December 31, 2024.
During
the fiscal years ended December 31, 2025 and December 31, 2024 neither the
Company nor anyone on its behalf consulted with EY regarding (i) the application of accounting principles to any specified transaction,
either completed or proposed, or the type of audit opinion that might be rendered on the Company’s financial statements, and neither
a written report nor oral advice was provided to the Company that EY concluded was an important factor considered by the Company in reaching
a decision as to any accounting, auditing, or financial reporting issue, or (ii) any matter that was either the subject of a “disagreement,”
as defined in Item 304(a)(1)(iv) of Regulation S-K, or a “reportable event,” as defined in Item 304(a)(1)(v) of Regulation
S-K.
The
following is a summary of the fees billed or expected to be billed to us by Kost Forer Gabbay & Kasierer, our independent
registered public accountants, for professional services rendered with respect to the fiscal years ended December 31, 2025, and 2024:
| | |
Kost
Forer Gabbay & Kasierer | |
| | |
2025 | | |
2024 | |
| Audit fees (1) | |
$ | 385,000 | | |
$ | 160,000 | |
| Audit-related fees (2) | |
| | | |
| | |
| Tax fees (3) | |
| | | |
| | |
| All other fees (4) | |
| 16,000 | | |
$ | - | |
| Total | |
$ | 391,000 | | |
$ | 160,000 | |
| |
(1) |
Audit
Fees. This category includes the fees related to the audit of our annual financial statements and the review of our interim quarterly
financial statements and services that are normally provided by our independent registered public accounting firm in connection with
its engagements for those years. This category also includes advice on audit and accounting matters that arose during, or as a result
of, the audit or the review of our interim financial statements. |
| |
|
|
| |
(2) |
Audit-Related
Fees. This category typically consists of assurance and related services by our independent registered public accounting firm
that are reasonably related to the performance of the audit or review of our financial statements and are not reported above under
“Audit Fees.” The services for the fees disclosed under this category include consents regarding equity issuances. |
| |
|
|
| |
(3) |
Tax
Fees. This category typically consists of professional services rendered by our independent registered public accounting firm
for tax compliance and tax advice. |
| |
|
|
| |
(4) |
All
Other Fees. This category includes aggregate fees billed in each of the last two fiscal years for Kost Forer Gabbay & Kasierer for products and services provided by the relevant independent registered public accounting
firm other than the services reported in the categories above. |
Pre-Approval
Policies and Procedures
Under
the Audit Committee’s pre-approval policies and procedures, the audit committee is required to pre-approve the audit and non-audit
services performed by our independent registered public accounting firm. On an annual basis, the Audit Committee pre-approves a list
of services that may be provided by the independent registered public accounting firm without obtaining specific pre-approval from the
audit committee. In addition, the Audit Committee sets pre-approved fee levels for each of the listed services. Any type of service that
is not included on the list of pre-approved services must be specifically approved by the Audit Committee or its designee. Any proposed
service that is included on the list of pre-approved services but will cause the pre-approved fee level to be exceeded will also require
specific pre-approval by the Audit Committee or its designee.
The
Audit Committee has delegated pre-approval authority to the Audit Committee chairman and any pre-approved actions by the Audit Committee
chairman as designee are reported to the Audit Committee for approval at its next scheduled meeting.
All
of the services rendered by Kost Forer Gabbay & Kasierer were pre-approved by the Audit Committee.
The
Board considered the audit fees, audit-related fees, tax fees and other fees paid to our accountants, as disclosed above, and determined
that the payment of such fees was compatible with maintaining the independence of the accountants.
ITEM
15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The
following documents are filed as part of this Annual Report on Form 10-K:
| |
(1) |
Financial
Statements: |
| |
(2) |
Financial
Statement Schedules: |
None.
See
“Index to Exhibits” for a description of our exhibits.
ITEM
16. FORM 10-K SUMMARY
None.
 |
Kost
Forer Gabbay & Kasierer
144
Menachem Begin Road, Building A,
Tel-Aviv
6492102, Israel
|
Tel:
+972-3-6232525
Fax: +972-3-5622555
ey.com
|
REPORT
OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To
the Shareholders and the Board of Directors of ENvue Medical, Inc.
Opinion
on the Financial Statements
We
have audited the accompanying consolidated balance sheets of ENvue Medical, Inc. (the Company) as of December 31, 2025 and 2024, the
related consolidated statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the two
years in the period ended December 31, 2025, and the related notes (collectively referred to as the “consolidated financial
statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial
position of the Company at December 31, 2025 and 2024, and the results of its operations and its cash flows for each of the two
years in the period ended December 31, 2025, in conformity with U.S. generally accepted accounting principles.
The
Company’s Ability to Continue as a Going Concern
The
accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed
in Note 2 to the financial statements, the Company has suffered recurring losses and negative cash flows from operations, and has stated
that substantial doubt exists about the Company’s ability to continue as a going concern. Management’s evaluation of the
events and conditions and management’s plans regarding these matters are also described in Note 2. The consolidated financial statements
do not include any adjustments that might result from the outcome of this uncertainty.
Basis
for Opinion
These
financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s
financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board
(United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities
laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We
conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain
reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company
is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits
we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion
on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our
audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error
or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding
the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant
estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits
provide a reasonable basis for our opinion.
Critical
Audit Matters
The
critical audit matters communicated below are matters arising from the current period audit of the financial statements that were communicated
or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the financial
statements and (2) involved our especially challenging, subjective or complex judgments. The communication of critical audit matters
does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the
critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they
relate.
Business Combination
|
| |
|
|
| Description
of the matter |
|
As described in Note 4 to the consolidated financial
statements, during 2025 the Company completed the acquisition of ENvue Medical Holding LLC (“ENvue”) for a total consideration
of $42,452 thousands, which included a combination of the Company’s ordinary shares, Series X Preferred Stock and Pre-Funded Warrants. The
transaction was accounted for as a business combination.
Auditing the Company’s accounting for its
acquisition of ENvue was complex due to the significant estimation uncertainty required by management in determining the fair value
of the identified intangible assets, which principally consisted of technology intangible asset in the amount of $3,500 thousands and
Customer relationships intangible asset in the amount of $1,820 thousands. The significant assumptions used to estimate the fair
value of the technology and customer relationships intangible assets included, among others, discount rate and projected revenue
growth rates. These significant assumptions are forward-looking and could be affected by future economic and market
conditions.
|
| |
|
|
| How
we addressed the matter in our audit |
|
To
test the estimated fair value of the technology and customer relationships intangible assets,
we performed audit procedures that included, among others, evaluating the methods and significant
assumptions used by the Company, and evaluating the completeness and accuracy of the underlying
data supporting the significant assumptions and estimates. We involved our valuation specialists
to assist in evaluation of the methodology used by the Company and certain assumptions included
in the fair value estimates. Our valuation specialists’ procedures included, among
others, developing a range of independent estimates for the discount rates used in the valuation
models and comparing those to the discount rates selected by management. In addition,
we evaluated the appropriateness of the related disclosures in relation to the acquisition.
|
| |
|
|
| Impairment of Goodwill and long-lived assets |
| |
|
|
| Description
of the matter |
|
As described in Note 21 to the consolidated financial
statements, during 2025 the Company recorded an impairment loss on goodwill and certain long-lived assets related to the ENvue reporting
unit. The Company evaluated the ENvue long-lived assets and goodwill for recoverability and determined that certain assets were not recoverable. As a result, the Company recognized a $645 thousands impairment loss related to ENvue long-lived assets, and a
$10,509 thousands impairment of goodwill. This represented the amount by which the carrying value exceeded the estimated fair value of these
assets.
Auditing the Company’s goodwill and long-lived assets impairment
assessments involved significant auditor judgment due to the significant estimation uncertainty in determining the fair value of the reporting
unit. The significant estimation uncertainty was primarily due to the sensitivity of the fair values to the significant underlying assumptions.
Significant assumptions used in the Company’s fair value estimate included discount rates, royalty rates, and certain assumptions
that form the basis of the forecasted results (e.g., revenue growth rates). These significant assumptions are especially challenging to
audit as they are forward looking and could be affected by future economic and market conditions.
|
| |
|
|
| How
we addressed the matter in our audit |
|
Our
audit procedures included, among others, testing the key inputs and significant assumptions discussed above. We involved our
valuation specialists to assist with our evaluation of discount rates and royalty rates. Additionally, we evaluated the significant
assumptions used by the Company, and validated the completeness and accuracy of the underlying data supporting the significant
assumptions and estimates. We also performed a sensitivity analysis of the significant assumptions to evaluate the change in the
fair values that would result from changes in the assumptions. |
/s/
KOST FORER GABBAY & KASIERER
A
Member of EY Global
We
have served as the Company’s auditor since 2025.
Tel-Aviv,
Israel
April
15, 2026
The
accompanying notes are an integral part of these consolidated financial statements
ENVUE
MEDICAL, INC.
Consolidated
Balance Sheets
(Amounts
in thousands except share and per share data)
| | |
December
31, 2025 | | |
December
31, 2024 | |
| ASSETS: | |
| | | |
| | |
| Current assets: | |
| | | |
| | |
| Cash and cash equivalents | |
$ | 4,224 | | |
$ | 752 | |
| Restricted cash | |
| 30 | | |
| - | |
| Trade receivables | |
| 289 | | |
| 98 | |
| Prepaid expenses and other accounts receivable | |
| 391 | | |
| 290 | |
| Inventory | |
| 2,337 | | |
| 2,191 | |
| Total current assets | |
| 7,271 | | |
| 3,331 | |
| | |
| | | |
| | |
| Non-current assets: | |
| | | |
| | |
| Property and equipment, net | |
| 119 | | |
| 9 | |
| Severance pay fund | |
| 125 | | |
| 173 | |
| Operating lease right-of-use assets | |
| 127 | | |
| 116 | |
| Long-term trade receivables | |
| 20 | | |
| - | |
| Intangible Assets, net | |
| 4,380 | | |
| - | |
| Goodwill | |
| 29,082 | | |
| - | |
| Total non-current assets | |
| 33,853 | | |
| 298 | |
| Total assets | |
$ | 41,124 | | |
$ | 3,629 | |
| | |
| | | |
| | |
| LIABILITIES AND STOCKHOLDERS’
EQUITY: | |
| | | |
| | |
| | |
| | | |
| | |
| Current liabilities: | |
| | | |
| | |
| Trade payables | |
$ | 719 | | |
$ | 47 | |
Arbitration liability | |
| 2,252 | | |
| 2,117 | |
| Other accounts payable and accrued expenses | |
| 2,345 | | |
| 491 | |
| Loans | |
| 1,080 | | |
| - | |
| Deferred revenue | |
| 175 | | |
| 15 | |
| Operating lease liabilities
- current | |
| 89 | | |
| 52 | |
| Total current liabilities | |
| 6,660 | | |
| 2,722 | |
| | |
| | | |
| | |
| Non-current liabilities: | |
| | | |
| | |
| Warrant liability | |
| 807 | | |
| - | |
| Accrued severance pay | |
| 132 | | |
| 216 | |
| Operating lease liabilities,
non-current | |
| 28 | | |
| 64 | |
| Total liabilities | |
$ | 7,627 | | |
$ | 3,002 | |
| | |
| | | |
| | |
| Commitments and contingencies | |
| - | | |
| | |
| | |
| | | |
| | |
| Stockholders’ equity [*]: | |
| | | |
| | |
| | |
| | | |
| | |
| Series G Preferred stock of $0.001 par value
- Authorized: 400,000 and 0 shares at December 31, 2025 and December 31, 2024, respectively; Issued and outstanding: 820 and 0 shares
at December 31, 2025 and December 31, 2024 | [*] |
| - | | |
| - | |
| Series H Preferred stock of $0.001 par value
- Authorized: 55,111 and 0 shares at December 31, 2025 and December 31, 2024, respectively; Issued and outstanding: 11,111 and 0 shares
at December 31, 2025 and December 31, 2024 | [*] |
| - | | |
| - | |
| Series X Preferred stock of $0.001
par value - Authorized: 62,220
and 0
shares at December 31, 2025 and December 31, 2024, respectively; Issued and outstanding: 53,100
and 0
shares at December 31, 2025 and December 31, 2024 | [*] |
| - | | |
| - | |
| Preferred stock value
| |
| - | | |
| - | |
| Common stock of $0.001 par value - Authorized:
40,000,000 shares at December 31, 2025 and December 31, 2024, respectively; Issued and outstanding: 1,100,413 and 37,894 shares at December
31, 2025 and December 31, 2024, respectively | [*] |
| 1 | | |
| 1 | |
| Additional paid in capital | [*] |
| 124,057 | | |
| 70,507 | |
| Accumulated other comprehensive income | [*] |
| (80 | ) | |
| (80 | ) |
| Accumulated deficit | [*] |
| (90,481 | ) | |
| (69,801 | ) |
| Total stockholders’
equity | [*] |
| 33,497 | | |
| 627 | |
| Total liabilities and
stockholders’ equity | |
$ | 41,124 | | |
$ | 3,629 | |
The
accompanying notes are an integral part of these consolidated financial statements
ENVUE
MEDICAL, INC.
Consolidated
Statements of Operations and Comprehensive Loss
(Amounts
in thousands except share and per share data)
| | |
| | |
| |
| | |
Year
Ended December 31, | |
| | |
2025 | | |
2024 | |
| | |
| | |
| |
| Revenues | |
$ | 2,553 | | |
$ | 2,558 | |
| Cost of revenues | |
| 2,400 | | |
| 1,050 | |
| Gross
profit | |
| 153 | | |
| 1,508 | |
| Operating expenses: | |
| | | |
| | |
| Research and development | |
| 1,762 | | |
| 909 | |
| Selling and marketing | |
| 2,493 | | |
| 720 | |
| General
and administrative | |
| 7,633 | | |
| 3,461 | |
| Impairment of goodwill and long-lived assets | |
| 11,154 | | |
| - | |
| | |
| | | |
| | |
| Total
operating expenses | |
| 23,042 | | |
| 5,090 | |
| | |
| | | |
| | |
| Loss from operations | |
| (22,889 | ) | |
| (3,582 | ) |
| | |
| | | |
| | |
| Financial
income (expense), net | |
| 4,397 | | |
| (104 | ) |
| | |
| | | |
| | |
| | |
| | | |
| | |
| Income
tax (expense) benefit | |
| 307 | | |
| (19 | ) |
| | |
| | | |
| | |
| Net
loss | |
$ | (18,185 | ) | |
$ | (3,705 | ) |
| | |
| | | |
| | |
| Preferred Stock dividends | |
| | | |
| | |
| Dividend on Series X Preferred Stock | |
| (1,700 | ) | |
| - | |
| Dividend on Series H Preferred Stock | |
| (396 | ) | |
| - | |
| Deemed dividend for down round feature on
Series H Preferred Stock | |
| (399 | ) | |
| - | |
| Deemed contribution on repurchase of Preferred
Series X | |
| 90 | | |
| - | |
| Deemed contribution on modification of Preferred
Series X | |
| 3,815 | | |
| - | |
| | |
| | | |
| - | |
| Net
loss available to common stockholders | |
| (16,775 | ) | |
| (3,705 | ) |
| | |
| | | |
| | |
| Basic and diluted net
loss available for holders of common stock [*] | |
$ | (26.69 | ) | |
$ | (137.30 | ) |
| | |
| | | |
| | |
| Weighted average common shares outstanding: | |
| | | |
| | |
| Basic
and diluted [*] | |
| 628,534 | | |
| 26,985 | |
| | |
| | | |
| | |
| Comprehensive
loss: | |
| | | |
| | |
| Net loss | |
$ | (18,185 | ) | |
$ | (3,705 | ) |
| Change
in foreign currency translation adjustments | |
| - | | |
| (13 | ) |
| Total
Comprehensive loss | |
$ | (18,185 | ) | |
$ | (3,718 | ) |
The
accompanying notes are an integral part of these consolidated financial statements
ENVUE
MEDICAL, INC.
Consolidated
Statement of Stockholders’ Equity
(Amounts
in thousands except share and per share data)
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
| |
| | |
Series
G Preferred Stock | | |
Series
X Preferred Stock | | |
Series
H Preferred Stock | | |
Common
Stock | | |
Additional Paid-in | | |
Accumulated Other
Comprehensive | | |
Accumulated | | |
Total Stockholders’ | |
| | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares | | |
Amount | | |
Shares [*] | | |
Amount [*] | | |
Capital | | |
Income | | |
Deficit | | |
Equity | |
| Balance, December 31, 2023 | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| - | | |
$ | - | | |
| 186,028 | | |
$ | 1 | | |
$ | 70,151 | | |
$ | (67 | ) | |
$ | (66,096 | ) | |
$ | 3,989 | |
| Stock-based compensation | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 356 | | |
| - | | |
| - | | |
| 356 | |
| Currency translation adjustment | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (13 | ) | |
| - | | |
| (13 | ) |
| Issuance of common stock upon exercise of pre-funded
warrants | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 19,291 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (3,705 | ) | |
| (3,705 | ) |
| Balance, December 31, 2024 | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 37,894 | | |
| 1 | | |
| 70,507 | | |
| (80 | ) | |
| (69,801 | ) | |
| 627 | |
| ENvue Merger | |
| - | | |
| - | | |
| 61,346 | | |
| - | | |
| - | | |
| - | | |
| 3,318 | | |
| - | | |
| 42,452 | | |
| - | | |
| - | | |
| 42,452 | |
| Exercise of warrants | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 5,000 | | |
| - | | |
| 102 | | |
| - | | |
| - | | |
| 102 | |
| Exercise of pre-funded warrants | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 250,734 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| Warrant exchange agreement | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 4,150 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| Issuance of Series G Preferred Stock | |
| 400,000 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 3,997 | | |
| - | | |
| - | | |
| 3,997 | |
| Conversion of Series G Preferred Stock into Common
Stock | |
| (399,180 | ) | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 709,419 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| Repurchase of Series X Preferred Stock | |
| - | | |
| - | | |
| (8,246 | ) | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (5,000 | ) | |
| - | | |
| - | | |
| (5,000 | ) |
| Dividend on Convertible Preferred Series X | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 1,700 | | |
| - | | |
| (1,700 | ) | |
| - | |
| Issuance of Series H Preferred Stock | |
| - | | |
| - | | |
| - | | |
| - | | |
| 11,111 | | |
| - | | |
| - | | |
| - | | |
| 6,602 | | |
| - | | |
| - | | |
| 6,602 | |
| Dividend on Convertible Preferred Series H | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| | | |
| - | | |
| - | | |
| 396 | | |
| - | | |
| (396 | ) | |
| - | |
| Issuance of Common Stock for private placement | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 74,114 | | |
| - | | |
| 1,835 | | |
| - | | |
| - | | |
| 1,835 | |
| Down round feature on Series H Preferred Stock | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 399 | | |
| - | | |
| (399 | ) | |
| - | |
| Reclassification of warrant liability to equity | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| | | |
| 1,067 | | |
| | | |
| - | | |
| 1,067 | |
| Rounding-up of fractional shares due to reverse stock
split | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| 15,785 | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | |
| Net loss | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| - | | |
| (18,185 | ) | |
| (18,185 | ) |
| Balance, December 31, 2025 | |
| 820 | | |
$ | - | | |
| 53,100 | | |
$ | - | | |
| 11,111 | | |
$ | - | | |
| 1,100,413 | | |
$ | 1 | | |
$ | 124,057 | | |
$ | (80 | ) | |
$ | (90,481 | ) | |
$ | 33,497 | |
| [*] |
Adjusted
to reflect the reverse stock splits (see note 11). |
The
accompanying notes are an integral part of these consolidated financial statements
ENVUE
MEDICAL, INC.
Consolidated
Statements of Cash Flows
(Amounts
in thousands except share and per share data)
| | |
| | |
| |
| | |
Year ended December 31, | |
| | |
2025 | | |
2024 | |
| Cash flows from operating activities: | |
| | | |
| | |
| Net loss | |
$ | (18,185 | ) | |
$ | (3,705 | ) |
| Adjustments to reconcile net loss to Net cash used in operating activities: | |
| | | |
| | |
| Depreciation and amortization | |
| 897 | | |
| 1 | |
| Stock-based compensation | |
| - | | |
| 356 | |
| Noncash interest expense | |
| 239 | | |
| 100 | |
| Other | |
| 19 | | |
| 1 | |
| Changes in deferred income taxes, net | |
| (373 | ) | |
| - | |
| Change in fair value of warrant liability | |
| (6,204 | ) | |
| - | |
| Impairment of goodwill and long-lived assets | |
| 11,154 | | |
| - | |
| Issuance cost allocated to warrant liability | |
| 1,420 | | |
| - | |
| Changes in operating assets and liabilities: | |
| | | |
| | |
| Trade receivable | |
| (179 | ) | |
| 221 | |
| Prepaid expenses and other accounts receivable | |
| (96 | ) | |
| (136 | ) |
| Inventory | |
| 249 | | |
| 541 | |
| Trade payables | |
| 552 | | |
| (91 | ) |
| Other accounts payable and accrued expenses | |
| 1,025 | | |
| 242 | |
| Deferred revenue | |
| 160 | | |
| (46 | ) |
| Operating right of use asset | |
| 71 | | |
| 34 | |
| Operating lease liabilities | |
| (85 | ) | |
| (34 | ) |
| Accrued severance pay, net | |
| (36 | ) | |
| - | |
| Net cash used in operating activities | |
| (9,372 | ) | |
| (2,516 | ) |
| | |
| | | |
| | |
| Cash flows from investing activities: | |
| | | |
| | |
| Cash acquired in Envue Merger | |
| 148 | | |
| - | |
| Purchase of property plant and equipment | |
| (60 | ) | |
| (3 | ) |
| Net cash provided by (used in) investing activities | |
| 88 | | |
| (3 | ) |
| | |
| | | |
| | |
| Cash flows from financing activities: | |
| | | |
| | |
| Proceeds from the issuance of common stock, preferred stock and warrants, net | |
| 19,089 | | |
| - | |
| Repurchase of Series X Preferred Stock | |
| (5,000 | ) | |
| - | |
| Proceeds from issuance of notes payable to related party | |
| 1,300 | | |
| - | |
| Payments of note payable to related party | |
| (1,300 | ) | |
| - | |
| Payments of short-term loan to related party | |
| (1,777 | ) | |
| - | |
| Proceeds from issuance of short-term loan payable | |
| 360 | | |
| - | |
| Proceeds from exercise of warrants | |
| 102 | | |
| 1 | |
| Other | |
| 12 | | |
| - | |
| Net cash provided by financing activities | |
| 12,786 | | |
| 1 | |
| | |
| | | |
| | |
| Effects of currency translation on cash and cash equivalents | |
| - | | |
| (13 | ) |
| | |
| | | |
| | |
| Net increase in cash, cash equivalents and restricted cash | |
| 3,502 | | |
| (2,531 | ) |
| Cash, cash equivalents and restricted cash at beginning of period | |
| 752 | | |
| 3,283 | |
| Cash, cash equivalents and restricted cash at end of period | |
$ | 4,254 | | |
$ | 752 | |
| | |
| | | |
| | |
| Reconciliation of cash, cash equivalents and restricted cash: | |
| | | |
| | |
| Cash and cash equivalents | |
| 4,224 | | |
| 752 | |
| Restricted cash at end of period | |
| 30 | | |
| - | |
| Total cash, cash equivalents and restricted cash | |
$ | 4,254 | | |
$ | 752 | |
| | |
| | | |
| | |
| Supplemental disclosures of cash flow information: | |
| | | |
| | |
| Cash paid for interest | |
$ | 25 | | |
$ | - | |
| Cash paid for taxes | |
$ | 52 | | |
$ | - | |
| Non-cash transactions: | |
| | | |
| | |
| Right of use asset obtained in the exchange for operating lease liabilities | |
$ | 24 | | |
$ | 145 | |
The
accompanying notes are an integral part of these consolidated financial statements
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
NOTE
1 - DESCRIPTION OF BUSINESS
ENvue
Medical, Inc. (formerly known as NanoVibronix, Inc.) (the “Company”) was incorporated as a Delaware corporation in
October 2003. The Company is a medical device company focusing on non-invasive biological response-activating devices that target
wound healing and pain therapy and can be administered at home without the assistance of medical professionals, utilizing its
proprietary low-intensity ultrasound (acoustic) technology. The Company’s principal research and development activities are
conducted in Israel through its wholly-owned subsidiary, NanoVibronix Ltd., a company registered in Israel, which commenced
operations in October 2003.
On
February 14, 2025, pursuant to the terms of that certain Agreement and Plan of Merger, dated as of February 14, 2025 (the “Merger
Agreement”), by and among the Company, NVEH Merger Sub, Inc., a Delaware corporation and a wholly-owned subsidiary of NVEH Merger
Sub I, Inc. (“First Merger Sub”), NVEH Merger Sub II, LLC, a Delaware limited liability company and a wholly-owned subsidiary
of the Company (“Second Merger Sub”), and ENvue Medical Holding Corp (“Predecessor ENvue”), the Company and Predecessor ENvue effected (i) a merger of First
Merger Sub with and into Predecessor ENvue, with First Merger Sub ceasing to exist and Predecessor ENvue becoming a wholly-owned subsidiary
of the Company, and (ii) the merger of Predecessor ENvue with and into Second Merger Sub (the “Second Merger” and, together
with the First Merger, the “ENvue Merger”), with Second Merger Sub being the surviving entity (the “Surviving Entity”).
At the effective time of the Second Merger, the certificate of formation of the Surviving Entity was amended and restated to, among other
things, change the name of the Surviving Entity to “ENvue Medical Holdings LLC.” See Note 4 – Merger.
ENvue
Medical Holdings LLC (formerly ENvue Medical Holding Corp.) (“ENvue”), a wholly-owned subsidiary of the Company, is a Delaware
limited liability company incorporated on June 5, 2024. ENvue has two wholly-owned subsidiaries: ENvue Medical (USA) Inc. and ENvue Medical
Ltd. (formerly ENvue Medical Israel Ltd.). ENvue is engaged in the research, development, marketing, and sale of medical devices in the
field of enteral feeding and is in the initial stage of commercializing its products.
NOTE
2 - LIQUIDITY AND PLAN OF OPERATIONS
As
of December 31, 2025, the Company has incurred recurring losses and negative cash flows from operations and has an accumulated deficit
of $90,481. For the twelve-month period
ended December 31, 2025, the Company used approximately $9,372 of cash in operations. The
Company’s ability to continue to operate is dependent mainly on its ability to successfully market and sell its products and
the receipt of additional financing until profitability is achieved.
The
Company expects to incur future net losses and its transition to profitability is dependent upon, among other things, the achievement
of a level of revenues adequate to support the cost structure. Until the Company achieves profitability or generates positive cash flows,
it will continue to be dependent on raising additional funds to fund its operations. The Company intends to fund its future operations
through cash on hand, additional private and/or public offerings of debt or equity securities or a combination of the foregoing. There
are no assurances, however, that the Company will be able to obtain an adequate level of financial resources that are required to fund
its operation.
The Company does not have sufficient resources to fund its operations for the next twelve months from the date of
the issuance of these consolidated financial statements. These
conditions raise substantial doubt about the Company’s ability to continue as a going concern. The accompanying consolidated financial statements
do not include any adjustments to reflect the possible future effects on recoverability and classification of assets or the amounts and
classification of liabilities that may result from the outcome of this uncertainty.
NOTE
3 - SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis
of presentation and principles of consolidation
The consolidated financial statements have been prepared
in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”).
The
accompanying consolidated financial statements include the accounts of ENvue Medical, Inc. (formerly known as NanoVibronix, Inc.) (the
“Company”) and its wholly-owned subsidiaries, including ENvue Medical Holdings LLC and its subsidiaries (collectively, “ENvue”)
from the date of the ENvue Merger (as defined herein), and NanoVibronix Ltd. All intercompany accounts and transactions have been eliminated
in consolidation.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Use
of estimates
The
preparation of the consolidated financial statements in conformity with U.S. GAAP requires management to make estimates, judgments and
assumptions. The Company believes that the estimates, judgments and assumptions used are reasonable based upon information available
at the time they are made. These estimates, judgments and assumptions can affect the reported amounts of assets and liabilities and disclosure
of contingent assets and liabilities at the dates of the financial statements, and the reported amounts of revenue and expenses during
the reporting period. Actual results could differ from those estimates.
Foreign
currency translation
The currency of the primary economic environment in which the operations
of the Company and its subsidiaries are conducted is the U.S. dollar; thus, the dollar is the functional currency of the Company and certain
subsidiaries.
Non-dollar
transactions and balances have been remeasured to dollars in accordance with ASC 830, “Foreign Currency Matters”. All transaction
gains and losses from remeasurement of monetary balance sheet items denominated in non-dollar currencies are reflected in the statements
of operations as financial income or expenses, as appropriate.
Cash
and cash equivalent
Cash
and cash equivalents consist of cash on hand and highly liquid investments with original maturities of three months or less, at the date
of purchase.
Concentration
of credit risk
Financial
instruments that potentially subject the Company to a concentration of credit risk consist of cash and cash equivalents, trade
receivables, and other accounts receivable. The Company holds cash and cash equivalents in various banking institutions. The
majority of the Company’s cash and cash equivalents and short-term bank deposits are invested with banks in United States.
Such investments are insured by the Federal Deposit Insurance Corporation (“FDIC”) up to $250.
Cash balances could exceed insured amounts at any given time. Generally, these investments may be redeemed upon demand, and the
Company believes that the financial institutions that hold the Company’s cash deposits are financially sound and, accordingly,
bear minimal risk. As of December 31, 2025, the Company had cash in excess of the FDIC insured amount totaling approximately $3,800.
The trade receivables of the Company are mainly derived
from sales to a diverse set of customers located primarily in the United States. The Company performs ongoing credit evaluations of its
customers and, to date, has not experienced any significant losses.
Trade
receivables
The
Company’s trade receivable balance consists of amounts due from its customers. Trade Receivables are recorded when the right
to consideration becomes unconditional. The Current Expected Credit Losses (“CECL”) impairment model requires an
estimate of expected credit losses, measured over the contractual life of an instrument, which considers forecasts of future
economic conditions in addition to information about past events and current conditions. Based on this model, the Company considers
many factors, including the age of the balance, collection history, and current economic trends. Credit losses are written off after
all collection efforts have ceased. Allowances for credit losses are recorded as a direct reduction from an asset’s amortized
cost basis. Credit losses and recoveries are recorded in selling, general and administrative expenses in the consolidated statements
of operations. Recoveries of financial assets previously written off are recorded when received. As of December 31, 2025 and 2024, the
credit losses allowance was immaterial.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Earnings
per share
The
Company computes net loss per share using the two-class method required for participating securities. The two-class method requires income
available to common stockholders for the period to be allocated between shares of Common Stock and participating securities based upon
their respective rights to receive dividends as if all income for the period had been distributed. The Company’s Series X Preferred
Shares and Series H Preferred shares would be entitled to dividends that would be distributed to the holders of Common Stock, based on
the conversion ratio, assuming conversion of all Convertible Series X Preferred Shares and Series H Preferred shares into shares of Common
Stock. The Company does not allocate losses to these participating securities as they do not share in the Company’s losses.
The
Company’s basic net loss per share is calculated by dividing net loss attributable to common by the
weighted-average number of shares, which include prefunded warrants, without consideration of potentially dilutive securities. The diluted
net loss per share is calculated by giving effect to all potentially dilutive securities outstanding for the period using the treasury
share method or the if-converted method based on the nature of such securities. Diluted net loss per share is the same as basic net loss
per share in periods when the effects of potentially dilutive shares of Common Stock are anti-dilutive.
Inventory
Inventories
are stated at the lower of cost or net realizable value. Net realizable value is the estimated selling prices in the ordinary course
of business, less reasonably predictable costs of completion, disposal, and transportation. Cost is determined using the “first-in,
first-out” method.
Inventory
write-offs are provided to cover risks arising from slow-moving items and obsolete items. The Company periodically evaluates
the quantities on hand relative to current and historical selling prices and historical and projected sales volume. Based on this evaluation,
provisions are made when required to write-down inventory to its net market value.
Goodwill
Goodwill
has been recorded as a result of the acquisition. Goodwill represents the excess of the purchase price in a business combination over
the fair value of identifiable tangible and intangible assets acquired. Goodwill is not amortized but rather is subject to an impairment
test.
ASC
No. 350, “Intangibles - Goodwill and other” (“ASC No. 350”) requires goodwill to be tested for impairment at
the reporting unit level at least annually or between annual tests in certain circumstances and written down when impaired.
ASC
No. 350 allows an entity to first assess qualitative factors to determine whether it is necessary to perform the quantitative goodwill
impairment test. If the qualitative assessment does not result in a more likely than not indication of impairment, no further impairment
testing is required. If it does result in a more likely than not indication of impairment, the quantitative goodwill impairment test
is performed. Alternatively, ASC No. 350 permits an entity to bypass the qualitative assessment for any reporting unit and proceed directly
to perform the quantitative goodwill impairment test. The Company performs the quantitative goodwill impairment test during the fourth
quarter of each fiscal year, or more frequently if impairment indicators are present and compares the fair value of the reporting unit
with its carrying value.
During the years ended December 31, 2025 and 2024, the Company recorded a goodwill impairment charge of
$10,509 and $0,
respectively. See Note 21.
Intangible
Assets
Purchased
intangible assets with finite lives are carried at cost, less accumulated amortization. Amortization is computed over the estimated
useful lives of the respective assets, which range from 5 to 7 years. Intangible assets, consisting primarily of
technology, tradename and trademarks and customer list, are amortized over their estimated useful lives on a straight-line basis or
in proportion to their economic benefits realized.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
The
estimated useful lives of the Company’s intangible assets are as follows:
SCHEDULE
OF INTANGIBLE ASSETS ESTIMATED USEFUL LIVES
| Intangible
Assets | |
Years | |
| Tradename and trademarks | |
| 5 | |
| Technology | |
| 7 | |
| Customer list | |
| 5 | |
Property
and equipment, net
Property
and equipment are stated at cost, net of accumulated depreciation. Depreciation is calculated using the straight-line method over the
estimated useful lives of the assets.
The
estimated useful lives of the Company’s property and equipment are as follows:
SCHEDULE
OF USEFUL LIVES OF THE COMPANY’S PROPERTY AND EQUIPMENT
| | |
Years | |
| Computers and peripheral equipment | |
| 3 | |
| Office furniture and equipment | |
| 5-7 | |
| Leasehold improvements | |
| The
shorter of term of the lease or the useful life of the asset | |
Impairment
of Long-Lived Assets
The
long-lived assets of the Company, including finite-lived intangible assets, are reviewed for impairment in accordance with ASC No. 360,
“Property, Plant and Equipment”, whenever events or changes in circumstances indicate that the carrying amount of an asset
may not be recoverable. The recoverability of assets to be held and used is measured by a comparison of the carrying amount of an assets
to the future undiscounted cash flows expected to be generated by the assets. If such review indicates that the carrying amount of long-lived assets is not recoverable, the carrying amount
of such assets is reduced to fair value.
During the years ended December 31, 2025 and 2024, the
Company recorded a long-lived asset impairment charge of $645 and $0, respectively. See Note 21.
Warrants
The Company accounts for warrants as either equity-classified or liability-classified
instruments based on an assessment of the warrant’s specific terms and applicable authoritative guidance. The assessment considers
whether the warrants are freestanding financial instruments, meet the definition of a liability under ASC 480, and meet all of the requirements
for equity classification, including whether the warrants are indexed to the Company’s own common stock and whether the warrants
meet the equity classification criteria. This assessment, which requires the use of professional judgment, is conducted at the time of
warrant issuance and as of each subsequent reporting period end date while the warrants are outstanding. Warrants that meet all the criteria
for equity classification, are required to be recorded as a component of additional paid-in capital. Warrants that do not meet all the
criteria for equity classification, are required to be recorded as liabilities at their initial fair value on the date of issuance and
remeasured to fair value at each balance sheet date thereafter. The liability-classified warrants are recorded under non-current liabilities.
Changes in the estimated fair value of the warrants are recognized in Financial income (expense), net in the consolidated statements of
operations.
The
Company issued warrants in connection with the Series G Preferred financing, which are classified as liabilities. Warrants issued in
connection with the Series H Preferred financing were initially classified as liabilities.
In October 2025, the Series H Warrants agreement was modified. As a result
of the modification, the Company determined that the Series H Warrants met the criteria for equity classification under ASC 815-40. Accordingly,
the Company reclassified the Series H Warrants from a liability to equity as of the modification date. All other outstanding warrants
are classified as equity.
Accrued
severance pay
The
Company’s liability for severance pay is for one Israeli employee and is calculated pursuant to Israeli Severance Pay Law
based on the most recent salary of the employees multiplied by the number of years of employment as of the balance sheet date and is
in large part covered by regular deposits with recognized pension funds, deposits with severance pay funds and purchases of
insurance policies. The value of these deposits and policies is recorded as an asset in the Company’s balance sheet.
Accrued severance pay liability on December 31, 2025 and 2024, was $132 and $216, respectively.
The majority of the Company’s
liability for severance pay is covered by the provisions of Section 14 of the Israeli Severance Pay Law (“Section 14”). Under
Section 14, employees are entitled to monthly deposits, contributed on their behalf to their insurance funds. Payments in accordance with
Section 14 release the Company from any future severance payments in respect of those employees. As a result, the Company does not recognize
any liability for severance pay due to these employees and the deposits under Section 14 are not recorded as an asset in the Company’s
consolidated balance sheet.
Additionally, the Company accrued severance costs
of $180,000 related to the departure of its former Chief Financial Officer. This amount is included within other accounts payable and
accrued expenses-(accrued payroll) on December 31, 2025 consolidated balance sheets.
Severance expense for the years ended
December 31, 2025, and 2024 amounted to $686 and $70, respectively.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Leases
The
Company determines if an arrangement is a lease at inception. The Company currently does not have any finance leases in which it is the lessee.
Operating lease right-of-use (“ROU”) assets and liabilities are recognized at the
present value of the future lease payments at the lease commencement date. The Company combines its lease payments and fixed payments
for non-lease components and account for them together as a single lease component. Operating lease ROU assets also include any prepaid
lease payments. Payments for variable lease costs are expensed as incurred and not included in the operating lease ROU assets and liabilities.
For short-term leases with a term of 12 months or less, operating lease ROU assets and liabilities are not recognized and the Company
records lease payments in the Consolidated Statements of Operations on a straight-line basis over the lease term.
The interest rate used to determine the present value of the future lease payments is the Company’s
incremental borrowing rate, because the interest rate implicit in the Company’s leases is not readily determinable. The Company’s incremental borrowing rate is estimated to approximate the interest rate on a collateralized
basis with similar terms and payments, and in economic environments where the leased asset is located. When determining lease terms, the
Company uses the non-cancellable period of the leases and do not assume renewals unless it is reasonably certain that the Company will
exercise that option. Operating lease expense is recognized on a straight-line basis over the lease term.
The Company’s lease terms may include options to extend or terminate the lease. These options are included
in the lease terms when it is reasonably certain they will be exercised.
The Company may sell its insertion systems to customers through bundled lease arrangements which typically include
insertion systems and nasoenteral tubes. Revenues under these bundled lease arrangements are allocated considering the relative standalone
selling prices of the lease and non-lease components included in the bundled arrangement. The primary accounting provision the Company
uses to classify transactions as sales-type or operating leases is whether the lease transfers ownership of the underlying asset to the
lessee by the end of the lease term. Systems included in arrangements meeting this condition are accounted for as sales-type leases and
revenue is recognized at lease commencement. When leases are determined to be operating leases, revenue is recognized over the term of the lease. For the year ended December 31, 2025, there
were no operating leases in which the Company is the lessor.
Revenue from sales-type leases is presented on a
gross basis when the Company enters into a lease to realize value from a product that it would otherwise sell in its ordinary course of
business. Interest income for the year ended December 31, 2025 was immaterial.
For the year ended December
31, 2025, the Company recognized $43 of revenue from sales-type lease agreements and $36 cost
of revenue. The Company’s short-term net investment in a lease receivable as of December 31, 2025 was $20 and
is presented within trade receivables in the consolidated balance sheets. The Company’s long -term net investment in a lease receivable
as of December 31, 2025 was $20 and is presented within long-term trade receivables in the consolidated balance sheets.
Revenue
recognition
Revenues
from product and services are recognized in accordance with ASC 606 “Revenue Recognition.” Five basic steps must be followed
before revenue can be recognized: (1) Identifying the contract(s) with a customer that create(s) enforceable rights and obligations;
(2) Identifying the performance obligations in the contract; (3) Determining
the transaction price, meaning the amount of consideration in a contract to which an entity expects to be entitled in exchange for transferring
promised goods or services to a customer; (4) Allocating the transaction price to the performance obligations in the contract, which
requires the company to allocate the transaction price to each performance obligation on the basis of the relative standalone selling
prices of each distinct performance obligation; and (5) Recognizing revenue when (or as) the entity satisfies a performance
obligation by transferring control over a promised good or service to a customer.
The Company’s performance obligation is generally
the sale and delivery of its products. Revenues from product sales is recorded at the transaction price, which includes estimates of variable
consideration that result from discounts as well as allowances for returns.
Regarding its NanoVibronix product sales,
revenue from product sales is recognized at a point in time when control of the product is transferred, which is generally upon
shipment to the customer.
Regarding
its ENvue product sales, the Company regularly sells its Systems and Nasoenteral tubes on a stand-alone basis and therefore
concludes these products are separate performance obligations. Revenue from product sales is recognized at a point in time when
control of the product is transferred, which is generally upon shipment to the customer.
When a contract includes one performance obligation, the entire transaction price is allocated to that performance
obligation. When a contract includes a combination of products and services, the transaction price is allocated to each performance obligation
based on its relative stand-alone selling price . The stand-alone selling prices are generally determined based on the prices at
which the Company separately sells the products and services. The Company’s contracts with its ENvue customers generally do not
include rights of return.
For customers of both NanoVibronix and ENvue, payments are typically due
between 30 and 60 days.
The
Company applied the practical expedient in ASC 606 and did not evaluate payment terms of one year or less for the existence of a significant
financing component. The related revenue is recognized net of any taxes collected from customers which are subsequently remitted to governmental
entities (e.g., sales tax and other indirect taxes). The Company elected to not disclose information about the remaining performance
obligations that have original expected durations of one year or less.
In
some of its contracts, the Company provides assurance warranty services to its customers, in accordance with legal provisions or
industry standards to ensure the quality of the products. As such, the Company recognizes a provision for warranties in its financial
statements as applicable. As of December 31, 2025, the Company’s provision for warranty amounted to $47.
Deferred revenue
During the year ended December 31,
2025, the Company entered into a purchase agreement with a customer for 1,000 units at $250
per unit (total contract value of $250).
The Company received a non-refundable advanced payment of $250
and delivered 300 units during the fourth quarter of 2025 recognizing $75
of revenue for the year. The remaining contract liability of $175
at December 31, 2025, representing 700 undelivered units, is classified as a current liability as the Company expects to fulfill the
remaining obligation within the next twelve months. During the year ended December 31, 2025, the Company recognized approximately
all of the revenue that was included in the current deferred revenues balance at the beginning of the period.
The following table presents the changes in the deferred
revenue for the year ended December 31, 2025:
SCHEDULE
OF DEFERRED REVENUE
| | |
| | |
| Balance as of December 31, 2024 | |
$ | 15 | |
| Payment | |
| 250 | |
| Revenue Recognized | |
| (90 | ) |
| Balance as of December 31, 2025 | |
$ | 175 | |
Income
taxes
The
Company accounts for income taxes in accordance with ASC 740, “Income Taxes”. This topic prescribes the use of the liability
method whereby deferred tax assets and liability account balances are determined based on differences between financial reporting and
tax bases of assets and liabilities and are measured using the enacted tax rates and laws that will be in effect when the differences
are expected to reverse. The Company provides valuation allowance, to reduce deferred tax assets to the amount that is more likely
than not to be realized.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
The
Company implements a two-step approach to recognize and measure uncertain tax positions. The first step is to evaluate the tax position
taken or expected to be taken in a tax return by determining if the weight of available evidence indicates that it is more likely than
not that, on an evaluation of the technical merits, the tax position will be sustained on audit, including resolution of any related
appeals or litigation processes. The second step is to measure the tax benefit as the largest amount that is more than 50% (cumulative
basis) likely to be realized upon ultimate settlement.
The
Company recognizes interest and penalties related to uncertain tax positions on the income tax expense line in the accompanying
consolidated statement of operations. Accrued interest and penalties are included on the related tax liability line in the
consolidated balance sheet. As of December 31, 2025 and 2024, the Company’s provision for uncertain tax positions was immaterial.
Stock-based
compensation
The Company accounts for share-based compensation
in accordance with ASC 718, “Compensation-Stock Compensation”. ASC 718 requires companies to estimate the fair value of share-based
payment awards on the date of grant using an option-pricing model. The value of the award is recognized as an expense over the requisite
service periods in the Company’s consolidated statement of operations. The Company recognizes compensation expenses for the value
of its awards with only service condition, which have graded vesting, based on the straight-line method over the requisite service period
of each of the awards. The Company accounts for forfeitures as they occur.
The
Company selected the Black-Scholes-Merton option pricing model as the most appropriate fair value method for its stock-options awards.
The option-pricing model requires a number of assumptions, of which the most significant are the expected stock price volatility and
the expected option term. Expected volatility was calculated based upon similar traded companies’ historical share price movements.
The expected option term represents the period that the Company’s stock options are expected to be outstanding. The Company currently
uses the simplified method and will continue to do so until sufficient historical exercise data supports using expected life assumptions.
The risk-free interest rate is based on the yield from U.S. Treasury zero-coupon bonds with an equivalent term. The expected dividend
yield assumption is based on the Company’s historical experience and expectation of no future dividend payouts. The Company has
historically not paid cash dividends and has no foreseeable plans to pay cash dividends in the future. During the
year ended December 31, 2025 no
stock-based compensation awards were granted.
Convertible
Debenture
The
Company accounted for its Convertible Debenture (see Note 13) under the fair value option pursuant to ASC 825. Under the fair value option, the Convertible Debenture is accounted for at fair value. Subsequent changes in fair value are recorded as a component of non-operating loss in the
consolidated statements of operations, except for adjustments related to instrument specific credit risk, which are recorded as
other comprehensive income. This election is made on an instrument-by-instrument basis as permitted under ASC 825. As a result of
electing the fair value option, direct costs and fees related to the Debenture are expensed as incurred.
The
Company’s Convertible Debenture was repaid in May 2025. During the year ended December 31, 2025, the Company recorded fair value
adjustment in the amounts of $22.
Variable
interest entities
The
Company evaluates its variable interests in variable interest entities (“VIEs”), and consolidates VIEs when the Company is
the primary beneficiary. The Company determines whether it is the primary beneficiary of a VIE based on its assessment of whether the
Company possesses both (i) the power to direct the activities that most significantly affect the VIE’s economic performance and
(ii) the obligation to absorb losses that could be significant to the VIE or the right to receive benefits that could be significant
to the VIE. The Company reevaluates the accounting for its VIEs upon the occurrence of events that could change the primary beneficiary
conclusion.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Business
combination
The
Company applies the provisions of ASC 805, “Business Combination” and allocates the fair value of purchase consideration
to the tangible assets acquired, liabilities assumed, and intangible assets acquired based on their estimated fair values. The excess
of the fair value of purchase consideration over the fair values of these identifiable assets and liabilities is recorded as goodwill.
Goodwill generated from a business combination is primarily attributable to synergies.
When
determining the fair values of assets acquired and liabilities assumed, management makes significant estimates and assumptions, especially
with respect to intangible assets. Significant estimates in valuing certain intangible assets include but are not limited to future expected
cash flows from acquired technology and acquired customer relationships from a market participant perspective, useful lives and discount
rates. Management’s estimates of fair value are based upon assumptions believed to be reasonable, but which are inherently uncertain
and unpredictable and, as a result, actual results may differ from estimates. During the measurement period, which may extend up to one year from the acquisition date, the Company may record
adjustments to the provisional fair values of the assets acquired and liabilities assumed, with a corresponding offset to goodwill. Upon
the earlier of the end of the measurement period or the final determination of the fair values of the assets acquired and liabilities
assumed, any subsequent adjustments are recorded in earnings.
Acquisition-related
expenses are recognized separately from the business combination and are expensed as incurred. See Note 4 Merger, for further
information.
Fair Value Measurements
Fair value is defined as the exchange price that would
be received from the sale of an asset or paid to transfer a liability in the principal or most advantageous market for the asset or liability
in an orderly transaction between market participants on the measurement date. The Company measures financial assets and liabilities at
fair value at each reporting period using a fair value hierarchy which requires the Company to maximize the use of observable inputs and
minimize the use of unobservable inputs when measuring fair value. A financial instrument’s classification within the fair value
hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Three levels of inputs may be used
to measure fair value:
Level 1 – Quoted prices in active markets for
identical assets or liabilities.
Level 2 – Inputs other than Level 1 that are
observable, either directly or indirectly, such as quoted prices for similar assets or liabilities; quoted prices in markets that are
not active; or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the
assets or liabilities.
Level 3 – Unobservable inputs that are supported
by little or no market activity and that are significant to the fair value of the assets or liabilities.
The carrying value of cash equivalents, restricted
cash, other accounts receivable, trade payables, and other accounts payable and accrued expenses approximate their fair value due to their
short-term maturities.
The Company warrant liabilities are measured at fair value using Level
3 inputs.
Recently
Adopted Accounting Pronouncement
In
December 2023, the Financial Accounting Standard Board (“FASB”) issued ASU 2023-09, Income Taxes (Topic 740),
Improvements to Income Tax Disclosures, which requires disaggregated information about the effective tax rate reconciliation as
well as information on income taxes paid. The Company adopted this guidance for the year ended December 31, 2025 on a prospective
basis. See Note 20 Income Taxes, for further information.
Recently issued accounting standards
In
November 2024, the FASB issued ASU 2024-03, Income Statement - Reporting Comprehensive Income - Expense Disaggregation Disclosure
(Subtopic 220-40), Disaggregation of Income Statement Expenses, which requires disclosure of disaggregated information about certain
expense captions presented in the Consolidated Statements of Operations as well as disclosure about selling expense. The guidance will
be effective for the Company for annual periods beginning January 1, 2027 and interim periods beginning January 1, 2028, with early adoption
permitted. It could be applied either prospectively or retrospectively. The Company is currently evaluating the impact on its financial
statement disclosures.
In
July 2025, the FASB issued Accounting Standards Update No. 2025-05, “Financial Instruments — Credit Losses (Topic 326): Measurement
of Credit Losses for Accounts Receivable and Contract Assets” (“ASU 2025-05”), which provides a practical expedient
for estimating expected credit losses for current accounts receivable and current contract assets. ASU 2025-05 will be effective for
annual periods beginning after December 15, 2025 and interim periods within those annual reporting periods and should be applied prospectively.
The Company is currently evaluating the impact of ASU 2025-05 on its consolidated financial statements and related disclosures.
In September 2025, the FASB issued ASU 2025-06, Intangibles
- Goodwill and Other - Internal-Use Software (Subtopic 350-40):Targeted Improvements to the Accounting for Internal-Use Software,
to modernize the accounting for costs related to internal-use software and better align it with current software development practices.
The amended guidance removes references to project stages and clarifies when entities are required to begin capitalizing eligible costs.
This guidance will be effective for the Company for annual periods beginning January 1, 2028, with early adoption permitted. The guidance
may be applied prospectively, retrospectively, or using a modified prospective transition method. The Company is currently evaluating
the impact of this ASU on its consolidated financial statements.
In May 2025, the FASB issued ASU
2025-03, Business Combinations (Topic 805) and Consolidation (Topic 810): Determining the Accounting Acquirer in the Acquisition
of a Variable Interest Entity, which clarifies current guidance for determining the accounting acquirer for a transaction
effected primarily by exchanging equity interests in which the legal acquiree is a variable interest entity that meets the
definition of a business. ASU 2025-03 is effective for annual reporting periods beginning after December 15, 2026, including interim
reporting periods within those annual reporting periods, with early adoption permitted. The Company is currently evaluating the
impact of this ASU on its consolidated financial statements.
NOTE
4 – MERGER
Agreement
and Plan of Merger
On
February 14, 2025, pursuant to the terms of that certain Agreement and Plan of Merger (as amended, restated, amended and restated, supplemented
or modified from time to time, the “Merger Agreement”), dated as of February 14, 2025, by and among us, NVEH Merger Sub I,
Inc., a Delaware corporation and a wholly-owned subsidiary of the Company (“First Merger Sub”), NVEH Merger Sub II, LLC,
a Delaware limited liability company and a wholly-owned subsidiary of the Company (“Second Merger Sub”), and ENvue Medical
Holdings, Corp. (“Predecessor ENvue” or “ENvue”), the Company and Predecessor ENvue effected (i) a merger of
First Merger Sub with and into Predecessor ENvue, with the First Merger Sub ceasing to exist and Predecessor ENvue becoming a wholly-owned
subsidiary the Company (the “First Merger”, and effective time of such First Merger, the “First Effective Time”)
and (ii) the merger of Predecessor ENvue with and into Second Merger Sub (the “Second Merger” and, together with the First
Merger, the “Merger”), with Second Merger Sub being the surviving entity of the Second Merger (“Surviving Entity”).
At the effective time of the Second Merger, the certificate of formation of the Surviving Entity was amended and restated to, among other
things, to change the name of the Surviving Entity to “ENvue Medical Holdings LLC.” In connection with the Merger Agreement,
the Company issued (i) 3,318
shares of common stock (the “Merger Shares”), which
such number of shares represented no more than 4.9%
(the “Exchange Cap”) of the outstanding shares of common stock as of immediately before the First Effective Time and (ii)
Pre-Funded Warrants to purchase up to 12,526
shares of our common stock (the “Merger Pre-Funded Warrants”)
at an exercise price of $0.001
per share, and (iii) 57,720
shares of Series X Non-Voting Convertible Preferred Stock (the
“Series X Preferred Stock”) in excess of the Exchange Cap to the holders of Predecessor ENvue in consideration for 100%
of Predecessor ENvue. In addition, the Company issued 3,626 shares
of Series X Preferred Stock to a service provider of ENvue, replacing its equity interest in ENvue, resulting in a total of 61,346 shares
of Series X Preferred Stock outstanding after the Merger.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
The Company concluded the merger resulted in the Company
obtaining a controlling financial interest in VIE in accordance with ASC 810, Consolidation. The Company determined that ENvue
was considered to be a VIE as it did not have sufficient equity to finance its activities without additional subordinated financial support.
The Company acquired all of the outstanding shares of ENvue and, therefore, is the sole equity holder and primary beneficiary. The Company
has the obligation to the absorb losses and right to receive the benefits of ENvue, and the power to direct the activities that most
significantly affect the economic performance of ENvue. Therefore, the Company is the primary beneficiary. Further, the Company concluded
that ENvue qualified as a business and accounted for the transaction as the acquisition of a business in accordance with ASC 805. As
the primary beneficiary, the Company was the acquirer in the transaction. During the measurement period, in 2025,
the Company revised certain aspects of the purchase price allocation. As a result, goodwill and additional paid-in capital increased
to reflect updated information available within the measurement period, in accordance with ASC 805, Business Combinations.
For
additional details regarding the Series X Preferred Stock, refer to note 11 – stockholders’ equity.
Given
that both companies operate in the medical device sector and demonstrate natural synergies and strategic alignment in their operations,
the merger was undertaken to combine and enhance their respective strengths.
The
acquisition date fair value of the consideration transferred amounted to $42,452, which included a combination of the Company ordinary
shares, Series X Preferred Stock and Pre-Funded Warrants. Issuance costs of Series X Preferred Stock were immaterial.
Acquisition-related
expenses of approximately $461 were expensed by the Company in general and administrative expenses in its condensed consolidated statements
of comprehensive loss for the year ended December 31, 2025.
The
transaction was accounted for as a business combination in accordance with ASC No. 805, “Business Combinations.” The total
purchase price was preliminarily allocated using information currently available to the Company and may be subject to change as additional
information is received during the respective measurement period, up to one year from the acquisition date. Preliminary allocation of
the purchase price to the tangible and identifiable intangible assets acquired and liabilities assumed based on their estimated fair
values as of the acquisition date is as follows:
SCHEDULE
OF INTANGIBLE ASSETS AND LIABILITIES ASSUMED BASED ON THEIR ESTIMATED FAIR
VALUES
| Purchase price: | |
| | |
| Common Stock and Pre-Funded Warrants
consideration | |
$ | 767 | |
| Series X Preferred Stock
consideration | |
| 41,685 | |
| Total purchase price | |
$ | 42,452 | |
| | |
| | |
| Allocated tangible assets: | |
| | |
| Net working capital | |
$ | (2,754 | ) |
| Property and equipment | |
| 112 | |
| Right-of-use assets | |
| 58 | |
| Lease liability | |
| (63 | ) |
| Deferred tax liability | |
| (373 | ) |
| Total
allocated tangible assets | |
$ | (3,019 | ) |
| | |
| | |
| Purchase price allocated
to intangible assets | |
| | |
| Tradename and trademarks | |
$ | 560 | |
| Proprietary technology | |
| 3,500 | |
| Customer relationships | |
| 1,820 | |
| Goodwill | |
$ | 39,591 | |
The
excess purchase price has been recorded as “goodwill” in the amount of $39,591.
The estimated useful life of the identifiable intangible assets is 5five
to seven years.
Goodwill is not expected to be tax deductible.
On the acquisition date, the Company considered the deferred tax impact of the excess fair value of the assets and
liabilities accounted for in the business combination over their historical cost basis. The Company recognized $373 of a deferred tax
liability which relates to the fair value of intangibles, other than goodwill and the fair value adjustments for the tangible assets acquired
over their historical cost basis. The Company will file a consolidated tax return in the U.S. together with ENvue and to utilize the benefit
of the Company’s loss carryforwards against the future taxable profit of ENvue and consequently decreased its valuation allowance
in an amount equal to the deferred tax liability recognized in the business combination.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Supplemental
Unaudited Pro Forma Information
Following
are the supplemental consolidated financial results of the Company and ENvue on an unaudited pro forma basis, as if the
acquisitions had been consummated as of the beginning of the fiscal year 2024 (i.e., January 1, 2024).
SCHEDULE OF SUPPLEMENTAL CONSOLIDATED FINANCIAL RESULT OF THE COMPANY
| |
|
Year Ended
|
| |
Year Ended | |
| |
|
December 31, 2025 |
| |
December
31, 2024 [*] | |
| Net revenue |
|
$ |
2,608 |
| |
$ | 2,923 | |
| Net loss |
|
$ |
(18,736 |
) | |
$ | (6,344 | ) |
The unaudited pro forma financial information below combines the historical
results of the Company with the historical results of ENvue for the year ended December 31, 2025 and 2024, as if the Merger had occurred
on January 1, 2024.
The unaudited pro forma financial information presented
is for informational purposes only and is not necessarily indicative of the results of operations that would have been achieved had the
Merger been completed at the beginning of fiscal year 2024, nor is it indicative of the future operating results of the combined company.
The pro forma results includes and adjustment related to purchase accounting, primarily amortization of acquisition-related intangible
assets.
NOTE
5 – PREPAID EXPENSES AND OTHER ACCOUNTS RECEIVABLES
Prepaid
expenses and other receivables consist of the following:
SCHEDULE OF PREPAID EXPENSES AND OTHER RECEIVABLES
| | |
2025 | | |
2024 | |
| | |
December
31, | |
| | |
2025 | | |
2024 | |
| | |
| | |
| |
| Prepaid expenses | |
$ | 137 | | |
$ | 120 | |
| Other receivables | |
| 254 | | |
| 170 | |
| | |
| | | |
| | |
| Total prepaid expenses and other accounts receivable | |
$ | 391 | | |
$ | 290 | |
NOTE
6 – INVENTORY
Inventory
consists of the following components:
SCHEDULE OF INVENTORY
| | |
| | | |
| | |
| | |
December 31, | |
| | |
2025 | | |
2024 | |
| | |
| | |
| |
| Raw materials | |
$ | 1,801 | | |
$ | 1,462 | |
| Finished goods | |
| 536 | | |
| 729 | |
| Total inventory | |
$ | 2,337 | | |
$ | 2,191 | |
Inventory
write-down charged to the cost of sales amounted to $346 and $0 for the year ended December 31, 2025 and 2024, respectively, to reduce
inventory to its net realizable value and for any excess or obsolete inventory.
The Company identified certain inaccuracies
in its 510(k) application for the PainShield MD Plus product and on August 19, 2025 submitted a request to the U.S. Food and Drug Administration
(FDA) to withdraw the clearance. While the Company is unaware of any safety issues related to the PainShield MD Plus product, it decided
to halt future sales. An appropriate inventory write-down was recorded for the impact of halt in sales in the amount of $137.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Note
7 – INTANGIBLE ASSETS, NET
The
following summarizes the Company’s intangible assets as of December 31, 2025:
SCHEDULE
OF INTANGIBLE ASSETS
| | |
Gross Carrying
Amount | | |
Accumulated
Amortization | | |
Impairment | | |
Balance as of
December 31, 2025 | |
| Tradename and trademarks | |
$ | 560 | | |
$ | (98 | ) | |
$ | (32 | ) | |
$ | 430 | |
| Proprietary technology | |
| 3,500 | | |
| (438 | ) | |
| (232 | ) | |
| 2,830 | |
| Customer relationships | |
| 1,820 | | |
| (319 | ) | |
| (381 | ) | |
| 1,120 | |
| | |
$ | 5,880 | | |
$ | (855 | ) | |
$ | (645 | ) | |
$ | 4,380 | |
For
the year ended December 31, 2025 amortization expense amounted to $855.
Expected
future amortization expense of intangible assets as of December 31, 2025 is as follows:
SCHEDULE
OF AMORTIZATION EXPENSE OF INTANGIBLE ASSETS
| | |
| | |
| 2026 | |
$ | 976 | |
| 2027 | |
| 976 | |
| 2028 | |
| 976 | |
| 2029 | |
| 622 | |
| 2030 | |
| 500 | |
| Thereafter | |
| 330 | |
| Total | |
$ | 4,380 | |
For the years ended December 31, 2025
and 2024, the Company recognized intangible asset impairment in the amount of $645 and $0, respectively. See Note 21 for further information.
Note
8 – GOODWILL
The
following table presents the changes in the carrying amount of goodwill, all of which was recognized in connection with the ENvue Merger and relates to the ENvue reporting unit, for the years ended December 31, 2025 and 2024:
SCHEDULE
OF GOODWILL
| Balance as of December 31, 2024 | |
$ | - | |
| Acquisitions | |
| 39,591 | |
| Impairment | |
| (10,509 | ) |
| Balance as of December 31, 2025 | |
$ | 29,082 | |
For
the years ended December 31, 2025 and 2024, the Company recorded goodwill impairment in the amounts of $10,509
and $0,
respectively. See Note 21 for further information.
Note
9 – OTHER ACCOUNTS PAYABLE AND ACCRUED EXPENSES
Other
accounts payable and accrued expenses consists of the following:
SCHEDULE OF OTHER ACCOUNTS
PAYABLE AND ACCRUED EXPENSES
| | |
| | | |
| | |
| | |
December 31, | |
| | |
2025 | | |
2024 | |
| R&D accrued expenses | |
$ | 625 | | |
$ | - | |
| Credit allowance | |
| 250 | | |
| - | |
| Compensation and payroll accrual | |
| 421 | | |
| 102 | |
| Taxes payable | |
| 117 | | |
| - | |
| Other accrued expenses | |
| 932 | | |
| 389 | |
| Total | |
$ | 2,345 | | |
$ | 491 | |
NOTE
10 – LEASES
The
Company has operating lease agreements with terms up to 3 years, including car and office space leases.
The components of operating lease costs were as follows:
SCHEDULE
OF OPERATING LEASE COSTS
| | |
| | |
| |
| | |
Year Ended December 31, | |
| | |
2025 | | |
2024 | |
| Fixed lease cost | |
$ | 124 | | |
$ | 61 | |
| Variable lease cost | |
| 3 | | |
| 1 | |
| | |
| | | |
| | |
| Total net lease costs | |
$ | 127 | | |
$ | 62 | |
The following table presents supplemental cash flows
information related to the lease costs for operating leases:
| |
|
Year
Ended
December 31, |
|
| |
|
2025 |
|
| Cash paid for amounts included
in measurement of lease liabilities: |
|
|
|
| Operating cash
flows for operating leases |
|
$ |
85 |
|
The
Company’s weighted-average remaining lease term relating to its operating leases is 2.21 years, with a weighted-average discount
rate of 10%.
The
following table presents information about the amount and timing of liabilities arising from the Company’s operating leases as
of December 31, 2025:
SCHEDULE OF LIABILITIES ARISING FROM OPERATING LEASES
| | |
| | |
| 2026 | |
$ | 93 | |
| 2027 | |
| 29 | |
| 2028 | |
| 5 | |
| Total undiscounted operating lease payments | |
| 127 | |
| Less: Imputed interest | |
| 10 | |
| Present value of operating
lease liabilities | |
$ | 117 | |
Sales-type Lease
Revenue from sales-type leases is presented on a gross basis when the Company
enters into a lease to realize value from a product that it would otherwise sell in its ordinary course of business. The Company’s
leases generally do not provide for a residual value guarantee. The Company’s lease arrangements are generally comprised of fixed
lease payments and do not include options to purchase the underlying assets and to extend or terminate the lease.
Interest income for the year ended December 31, 2025
was immaterial.
For the year ended December
31, 2025, the Company recognized $43 of revenue from sales-type lease agreements and $36 cost of revenue. The Company’s short -term
net investment in a lease receivable as of December 31, 2025 was $20 and is presented within trade receivables in the consolidated balance
sheets. The Company’s long -term net investment in a lease receivable as of December 31, 2025 was $20 and is presented within long-term
trade receivables in the consolidated balance sheets.
The following table illustrates
the Group’s future sales-type lease receipts as of December 31, 2025:
SCHEDULE
OF SALES-TYPE LEASE
| | |
| | |
| Year ending December 31, | |
| |
| 2026 | |
$ | 20 | |
| 2027 | |
| 20 | |
| Total future minimum receipts | |
$ | 40 | |
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
NOTE
11 - STOCKHOLDERS’ EQUITY
Common
Stock
The
common stock confers upon the holders the right to receive notice to participate and vote in general meetings of the Company, and the
right to receive dividends, if declared, and to participate in the distribution of the surplus assets and funds of the Company in the
event of liquidation, dissolution or winding up of the Company.
Reverse
stock splits
On
March 13, 2025, the Company effected a 1-for-11 reverse stock split (the “March 2025 Reverse Stock Split”). On August 11,
2025, the Company effected a 1-for-10 reverse stock split (the “August 2025 Reverse Stock Split” and together with the March 2025 Reverse Stock Split, (the “Reverse Stock Splits”).
As
a result of the March 2025 Reverse Stock Split, every eleven (11) shares of issued and outstanding Common Stock will be automatically
combined into one (1) issued and outstanding share of Common Stock, without any change in the par value per share.
As
a result of the August 2025 Reverse Stock Split, every ten (10) shares of issued and outstanding Common Stock will be automatically combined
into one (1) issued and outstanding share of Common Stock, without any change in the par value per share. No fractional shares were issued
as a result of the Reverse Stock Splits.
Any
fractional shares that would otherwise have resulted from the Reverse Stock Splits were rounded up to the next whole number. The number
of authorized shares of common stock under the Company’s Amended and Restated Certificate of Incorporation, as amended, remained
unchanged at 40,000,000 shares.
All
references in these consolidated financial statements to the number of shares, price per share and weighted average number of shares of common stock outstanding
prior to the Reverse Stock Splits have been adjusted to reflect the Reverse Stock Splits on a retroactive basis, unless otherwise noted.
Underwritten
Public Offering, Series G Convertible Preferred Stock
On
May 15, 2025, the Company announced the closing of an underwritten public offering (the “2025 Underwritten Offering”) of 400,000
shares of the Company’s Series G Convertible Preferred Stock (“Series G Preferred Stock”), with a par value $0.01
per share, and liability classified warrants to purchase up to 490,198
shares of common stock, par value $0.01
per share of the Company at an exercise price of $20.40
per share (the “May 2025 Warrants”). The combined public offering price of each share of Series G Preferred Stock
together with an accompanying May 2025 Warrant was $25.
The May 2025 Warrants have a term of five years from the initial issuance date and are exercisable immediately upon issuance. The
Company also issued warrants (the “May 2025 Representative’s Warrants”) to purchase up to 24,510
shares of common stock with an exercise price of $20.40 per share as issuance costs. The May 2025 Representative’s Warrants
expire five years from the date of commencement of sales in the 2025 Underwritten Offering.
On
May 15, 2025, prior to the closing of the 2025 Underwritten Offering, the Company filed the Certificate of Preferences, Rights and Limitations
of the Series G Convertible Preferred Stock (the “Series G Certificate of Designations”) with the Secretary of State of the
State of Delaware, which became effective upon filing. Pursuant to the terms of the Series G Certificate of Designations, the holders
of the Series G Preferred Stock are entitled to receive cumulative dividends at the rate per share of 9%
per annum of the stated value per share until the fifth anniversary of the date of issuance of the Series G Preferred Stock, which such
dividends may be paid, at the Company’s option, in up to an aggregate of 220,588
shares of common stock. In addition, in accordance with the
Series G Certificate of Designations, accrued and unpaid dividends are payable upon the conversion of the Series G Preferred Stock prior
to the fifth anniversary of issuance, upon any liquidation, dissolution, or winding up of the Company, and in connection with certain
fundamental transactions. The five year 9%
per annum dividend will be paid upon conversion of the Series G Preferred Stock irrespective of the timing of conversion such that upon
conversion, the conversion price will incorporate the five-year 9%
dividend.
The
aggregate net proceeds of the 2025 Underwritten Offering were approximately $8,200, after deducting approximately $1,800 of underwriting discounts, commissions and other offering costs and expenses.
Conversion of Series G Convertible Preferred Stock
Between May 16, 2025, and July 1, 2025, a majority
of the Series G Preferred Stockholders exercised their conversion right, converting 399,180 shares of Series G Preferred Stock into 709,419
shares of Common Stock. For the year ended December 31, 2025, there were 820 outstanding shares of Series G Preferred Stock.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Series
X Non-Voting Convertible Preferred Stock
On
February 14, 2025, in connection with the Merger (see Note 4), the Company issued 57,720
shares of Series X Non-Voting Convertible Preferred Stock (the “Series X Preferred Stock”) to the holders of Predecessor
ENvue. In addition, the Company issued 3,626 shares
of Series X Preferred Stock to a service provider of ENvue, replacing its equity interest in ENvue, resulting in a total of 61,346 shares
of Series X Preferred Stock outstanding after the Merger.
The
Merger was consummated and completed on February 14, 2025.
After
giving effect to the Merger, pursuant to the terms and conditions of the Merger Agreement: (i) the holders of the outstanding equity
of Predecessor ENvue immediately prior to the effective time of the First Merger (“First Effective Time”) own 19.9% of the
common stock of the Company and 85.0% of the outstanding equity of the Company (assuming the Series X Preferred Stock is converting at
a ratio of 100:1) immediately following the First Effective Time, which following stockholder approval will allow the Series X Preferred
Stock to convert to common stock of the Company which may result in the holders of Predecessor ENvue to own 85% of the common stock of
the Company, and (ii) the holders of our outstanding equity immediately prior to the First Effective Time own 80.1% of the common stock
of the Company and 15.0% of the outstanding equity of the Company (assuming the Series X Preferred Stock is converting at a ratio of
100:1) immediately following the First Effective Time, which following stockholder approval which will allow the Series X Preferred Stock
to convert to common stock of the Company which may result in our holders owning 15% of common stock of the Company.
Each
share of Series X Preferred Stock has a stated value of $606.3756 and
ranks senior to the Company’s common stock with respect to dividend rights and rights upon liquidation, dissolution or winding
up. Upon liquidation, prior to stockholder approval, the Series X Preferred holders shall be entitled to receive the greater of (i)
twice the aggregate stated value of the preferred shares and (ii) the amount the Holder would be entitled to receive if the
preferred shares were fully converted. The Series X Preferred Stock is not subject to mandatory redemption and may not be redeemed
at the option of the Company or the holder. For the year ended December 31, 2025 and 2024, 53,100
and 0 shares
of Series X Preferred Stock, respectively, were issued and outstanding. On July 22, 2025, the Company repurchased 8,246
outstanding shares of its Series X Preferred Stock from Alpha for $5
million from the use of proceeds from the July Private Placement.
Holders
shall be entitled to receive, and the Company shall pay, dividends on shares of Series X Preferred Stock, based on the Stated Value,
at a rate of eight percent (8%) per annum, commencing on the three (3) month anniversary of the Original Issue Date (as defined in the
Series X Certificate of Designations) until the date the Company obtains the Stockholder Approval. Such dividends can be paid in the
form of cash or additional issuances of shares of Series X Preferred Stock based on the Stated Value, with such type of payment determined
in the sole discretion of the Company, and accrue and be compounded daily on the basis of a 360-day year and twelve (12) 30-day months
and shall be paid the earlier of: (i) promptly after conversion of the Series X Preferred Stock or (ii) quarterly starting on the six
(6) month anniversary of the Original Issue Date. For the year ended December 31, 2025, the Company recorded $1,874,
of dividends attributable to Series X Preferred stockholders,
respectively.
On
May 12, 2025, the Company filed the Series X Certificate of Amendment with the Secretary of State of the State of Delaware, thereby amending
the Series X Certificate of Designations. The Series X Certificate of Amendment became effective with the Secretary of State of the State
of Delaware upon filing. The amendment decreased the Series X Preferred Stock conversion price from $66.69 to $20.40.
The
Company concluded that the modification of the Series X Preferred stock should be accounted for as an extinguishment. As such, the difference
between the fair value of the modified Series X Preferred Stock and their carrying amount was accounted for as a deemed contribution
in the amount of $3,815.
In conjunction with the July Purchase Agreement (as defined below), on July 22, 2025, the Company repurchased 8,246
outstanding shares of its Series X Preferred Stock from one investor in accordance with the terms of the Certificate of Designations
of the Series X Preferred Stock for $
5
million from the use of proceeds from the July Private Placement. The repurchase resulted in a deemed contribution of
$90.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Private
Placement, Series H Convertible Preferred Stock
On
July 18, 2025, the Company entered into a Securities Purchase Agreement (the “July 2025 Purchase Agreement”) with a certain
institutional investor (the “July 2025 Investor”), pursuant to which it agreed to sell to the Investor (i) an aggregate of
8,889 shares of the Company’s newly-designated Series H Convertible Preferred Stock (“Series H Preferred Stock”), and
(ii) warrants to acquire up to an aggregate of 467,836 shares of common stock (the “July 2025 Warrants”) at an exercise price
of $22.50 (the “July 2025 Private Placement” and such closing, the “Initial Closing”).
Pursuant
to the terms of the July 2025 Purchase Agreement, the Company also agreed to issue 2,222 shares of Series H Preferred Stock and warrants
to purchase up to 116,960 shares of common stock at an exercise price of $22.50 in a second closing (the “Second Closing”),
subject to the satisfaction of customary closing conditions. Additionally, pursuant to the terms of the July 2025 Purchase Agreement,
the Company has agreed that during the period ending 36 months from the effective date of the registration statement (the “Resale
Registration Statement”) registering the resale of the shares of common stock underlying the Series H Preferred Stock and the July
2025 Warrants, the July 2025 Investor shall have the right to purchase up to 44,000 additional shares of Series H Preferred Stock.
The
Initial Closing and occurred on July 22, 2025, and the Second Closing occurred October 30, 2025. The aggregate net proceeds from the
July 2025 Private Placement was approximately $9,000,
after deducting placement agent fees and other offering expenses payable by the Company of $958.
The Series H Preferred Stock and the option to purchase additional shares of Series H Preferred Stock were classified as equity,
while the warrants and the obligation to issue additional warrants and Series H Preferred Stock were classified as liability. The
Company allocated the financing proceeds to the freestanding financial instruments. Thus, the Company allocated $6,601 to
equity and $2,772 to
warrant liability. As a result of amendment on November 1, 2025, the Company reclassified the warrant liability to equity.
Upon any liquidation, dissolution or winding-up of
the Corporation, whether voluntary or involuntary, the Holders shall be entitled to receive out of the assets, whether capital or surplus,
of the Corporation an amount equal to the Stated Value, plus any accrued and unpaid dividends thereon.
On January 30, 2026, the Company entered into that
certain Amendment Agreement (the “Series H Amendment Agreement”) with the Required Holders (as defined in the Series H Amendment
Agreement) to remove the Floor Price (as defined in the Series H Certificate of Designations) in consideration of the holders of the Series
H Preferred Stock exercising $2,500,000 of the Additional Investment Right (as such concept is described in the Series H Purchase Agreement
by and between us and the holders of the Series H Preferred Stock. See Note 22 – Subsequent Events.
Holders
of the Series H Preferred Stock shall be entitled to receive cumulative dividends at the rate per share (as a percentage of the stated
value per share) of 9%
per annum, payable on each conversion date of the Series H Preferred Stock in duly authorized, validly issued, fully paid and non-assessable
shares of common stock at the conversion price then in effect. The stated value of the Series H Preferred Stock is $1,000
per share. The Company recorded $403
of dividends attributable to Series H Preferred stockholders
for the year ended December 31. 2025
September
2025 Registered Direct Offering
On
September 16, 2025, the Company entered into a securities purchase agreement with a single institutional investor, pursuant to which
the Company issued and sold (i) 74,114 shares of common stock and (ii) prefunded warrants to purchase up to 217,090 shares of common
stock (the “September 2025 Pre-Funded Warrants”) pursuant to an effective shelf registration statement on Form S-3 (the “September
2025 Offering”). The offering price was $7.01 per share of common stock and $7.009 per September 2025 Pre-Funded Warrant, which
is the price of each share of common stock sold in the September 2025 Offering, minus the $0.001 exercise price per September 2025 Pre-Funded
Warrant. The net proceeds from the September 2025 Offering were approximately $1,880, after deducting placement agent fees and
other offering expenses of $163.
Down-round Feature
As the offering price of the registered direct offering
was less than the conversion price of Series H Preferred Stock, pursuant to Series H Preferred Stock terms, a down-round feature was triggered,
resulting in the conversion price of Series H Preferred Stock decreasing to $7.01. As a result of the down-round feature trigger, the
Company recorded deemed dividend of $399.
Share-based
compensation
On
December 19, 2024, stockholders approved the NanoVibronix, Inc. 2024 Long-Term Incentive Plan (the “2024 Plan”), as a successor
to the NanoVibronix 2014 Long-Term Incentive Plan, which was adopted by the Board on November 6, 2023. As of December 31, 2024, under
the 2024 Plan, 60,000 shares of the Company’s common stock were reserved for issuance. On March 14, 2025, the Company effected the 2025 Reverse
Stock Split. Consequently, the number of shares of common stock of the Company reserved for issuance pursuant to awards under the 2024
Plan was reduced to 5,455 shares. As of December 31, 2025, there were 1,044 shares of common stock available to be issued under the plan.
During
the year ended December 31, 2025, no employee options were exercised, no options were granted and 95 options expired.
During
the year ended December 31, 2025 and 2024, stock-based compensation expense related to these options were approximately $0 and $356,
respectively.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
The following is a
summary of options activity for the periods presented:
SCHEDULE OF OPTIONS ACTIVITY
| | |
Shares
Under Options | | |
Weighted
Average Exercise Price per Share | | |
Weighted
Average Remaining Life (Years) | |
| Outstanding – December 31, 2024 | |
| 4,506 | | |
| 35 | | |
| 9.24 | |
| Granted | |
| - | | |
| - | | |
| - | |
| Exercised | |
| - | | |
| - | | |
| - | |
| Expired | |
| (95 | ) | |
| 514 | | |
| - | |
| Outstanding – December 31, 2025 | |
| 4,411 | | |
| 272 | | |
| 8.43 | |
Warrants
On
August 30, 2023, the Company issued (a) Pre-Funded Warrants to purchase up to 26,427
shares of Common Stock with an exercise price of $0.001
per share, (b) Series A-1 Warrants to purchase up to 26,427
shares of Common Stock with an exercise price of $161.70
per share (the “Series A-1 Warrants”) and (c) Series A-2 Warrants to purchase up to 26,427
shares of Common Stock with an exercise price of $161.70
per share (the “Series A-2 Warrants”), or a total of 77,644
warrants, in conjunction with a private placement by and between the Company and an institutional investor, pursuant to a securities
purchase agreement dated August 30, 2023 (the “August 2023 Private Placement”). The Series A-1 Warrants are exercisable
immediately upon issuance and expire on March 1, 2029. The Series A-1 Warrants were exchanged, described below, and the Series A-2
Warrants expired on October 1, 2024.
Warrant
Exchange Agreement
On
January 7, 2025, the Company entered into a securities exchange agreement (the “Exchange Agreement”) with a certain
institutional investor pursuant to which the Company agreed to issue an aggregate of (i) 4,149
shares of common stock (the “January 2025 Exchange Shares”), (ii) a warrant to purchase up to 15,856
shares of common stock (the “January 2025 Warrant”), and (iii) a pre-funded warrant to purchase up to 17,813
shares of common stock (the “January 2025 Pre-Funded Warrant”), in exchange for the Series A-1 Warrant held by the
Holder to purchase up to 26,427
shares of common stock at an exercise price of $161.70
per share (the “Exchange”). As a result of the Exchange the Company cancelled the Series A-1 Warrant. The January 2025
Warrant has an exercise price of $68.296
per share.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Subsequent
to the Exchange, the January pre-funded warrants were exercised in full on a cashless basis for an aggregate of 22,835 shares
of common stock. In addition, the January 2025 warrants were also exercised in full on a cashless basis for an aggregate of 9,151 shares
of common stock.
The
following is a summary of warrant activity for the periods presented:
SCHEDULE OF WARRANT ACTIVITY
| | |
Warrants | | |
Weighted
Average
Exercise
Price
per
Share | | |
Weighted
Average
Remaining
Life
(Years) | |
| Outstanding – January 1, 2024 | |
| 78,484 | | |
$ | 116.53 | | |
| 3.60 | |
| Granted | |
| - | | |
| - | | |
| - | |
| Exercised | |
| (19,291 | ) | |
$ | 0.01 | | |
| - | |
| Cancelled | |
| (26,537 | ) | |
| 163.07 | | |
| - | |
| Outstanding – December 31, 2024 | |
| 32,656 | | |
$ | 147.04 | | |
| 2.51 | |
| Granted | |
| 1,411,288 | | |
| 18.30 | | |
| 2.61 | |
| Exercised | |
| (271,931 | ) | |
| 4.36 | | |
| - | |
| Cancelled | |
| (26,428 | ) | |
| 14.70 | | |
| - | |
| Outstanding – December 31, 2025 | |
| 1,145,585 | | |
$ | 21.51 | | |
| 2.57 | |
NOTE
12– WARRANT LIABILITY
The May 2025 Warrants (see note 11) do not meet all the equity classification criteria and therefore
were determined to be classified as liabilities measured at fair value through earnings. The Company utilized the Black Scholes Model
to calculate the value of the May 2025 Warrants issued during the year ended December 31, 2025.
The
fair value of the May 2025 Warrants was $5,306 was estimated at the date
of issuance using the following assumptions: dividend yield 0%; expected term of 5.0
years; equity volatility of 145.65%; and a risk-free
interest rate of 4.06%.
The aggregate
fair value of the May 2025 Warrants was $807 and
was estimated on December 31, 2025 utilizing the Black Scholes Model and using the following assumptions: stock price $2.31, exercise
price $20.40, dividend yield 0%; remaining term of 4.38 years;
equity volatility of 149.77%; and a risk-free interest rate of 3.73%.
The
fair value of the July 2025 Warrants of $2,772 was estimated at the date of issuance using the following assumptions: dividend yield
0%; expected term of 1.5 years; equity volatility of 170.97%; and a risk-free interest rate of 3.94%.
During October 2025, the July 2025 Warrants were
modified and as a result of the modification, the Company concluded that the July 2025 Warrants were no longer required to be classified
as a liability under ASC 815-40. As such, the Company reclassified the July 2025 Warrants liability to equity as of the modification date.
The fair value of the July 2025 Warrants of $1,067
was estimated at the modification date using the following assumptions: dividend yield 0%; expected term of 1.22 years; equity volatility
of 184%; and a risk-free interest rate of 3.59%.
The
Company recognized changes in the fair value of the warrant liability of $6,204
during the year ended December 31, 2025, as financial income
on the consolidated statement of operations.
Changes
in the warrant liability balance
Below
is the change in the warrant liability balance for the year ended December 31, 2025:
SCHEDULE
OF CHANGE IN WARRANT LIABILITY
| Balance as of December 31, 2024 | |
$ | - | |
| Issuance | |
| 8,078 | |
| Reclassification of warrant liability to equity | |
| (1,067 | ) |
| Change in fair value | |
| (6,204 | ) |
| Balance as of December
31, 2025 | |
$ | 807 | |
See
note 17 for assets and liabilities which are measured at fair value on a recurring basis by level within the fair value hierarchy.
NOTE
13 – CONVERTIBLE DEBENTURE
On
February 13, 2025, the Company entered into a Securities Purchase Agreement (the “PIPE Purchase Agreement”) with Alpha Capital Anstalt (“Alpha”), pursuant to which the Company sold in a private placement senior convertible debenture (the “Debenture”)
due the earlier of (i) the date that is the 30-day anniversary of the effective date of stockholder approval of the issuance of the shares
of common stock upon the conversion of the debentures and (ii) November 13, 2025 (the date that is nine months following the date of
issuance of the Debenture) (“Maturity Date”), having an aggregate principal amount (the “Principal Amount”) of
$500 (the “Debenture Transaction”). The closing of the Debenture Transaction occurred on February 14, 2025.
On
March 26, 2025, the Company amended and restated the Debenture (the “A&R Debenture”) to increase the Principal Amount
to $1,300.
On
the Maturity Date, the Company shall pay Alpha in cash or, at the option of Alpha, in the form of conversion shares, or
a combination thereof, the entire outstanding Principal Amount of the A&R Debenture, together with accrued and unpaid interest thereon,
the applicable exit fee and any other amounts due thereunder. Following the receipt of Debenture Stockholder Approval, the A&R Debenture
shall be convertible, in whole or in part, into shares of common stock, at the option of Alpha, at the initial conversion price
of $48.906 (the “Conversion Price”), which is subject to customary anti-dilution adjustments, and which such Conversion Price
shall not be lower than the floor price of $9.7812. The A&R Debenture bears interest at the rate of 8.0% per annum, payable on the
Maturity Date.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
The
Company elected to account for the A&R Debenture and all the embedded features under the fair value option. Subsequent changes in
fair value are recorded in financial income (expense), net in the consolidated statements of operations. As a result of electing the
fair value option, direct costs and fees related to the Debenture were expensed as incurred.
For
the year ended December 31, 2025, the Company recorded a loss of $22, related to the change in fair value of the A&R Debenture which
was recognized in other income (expense) in the consolidated statements of operations. On May 19, 2025, the Company repaid the A&R
Debenture in full.
The
following table presents the change in the balance of the A&R Debenture for periods identified:
SCHEDULE
OF A&R DEBENTURE
| | |
Year Ended
December 31, 2025 | |
| Balance, beginning of period | |
$ | - | |
| Proceeds received | |
| 1,300 | |
| Change in fair value | |
| 22 | |
| Payments on debenture | |
| (1,322 | ) |
| Balance, end of period | |
| - | |
NOTE
14 – LOANS
ENvue
Consolidated Secured Note
On
January 17, 2025, ENvue issued a Consolidated Secured Note (as amended, the “Alpha Note”) in the aggregate principal
amount of $2,497
to Alpha, which such Alpha Note was funded in several tranches. The Alpha Note does not bear interest and is secured by Collateral
(as defined in the ENvue Note). The aggregate principal amount owed under the Alpha Note shall be due and payable on the earlier of
(i) the receipt of shareholder approval by the Company of the Parent Stockholder Matters (as defined in that certain Merger
Agreement) and (ii) December 31, 2025. The Alpha Note could be paid in whole or in part by ENvue at any time, at the option of
ENvue. As part of the acquisition accounting, the Company recorded the loan at its fair value of $2,258.
The difference between the face value and the fair value will be recognized as interest expense over the life of the note. See also
Note 22.
For
the year ended December 31, 2025, the Company recognized $239, respectively, of amortized interest expense related to the note. During
the year ended December 31, 2025, the Company paid $1,417, toward redeeming the Alpha Note, leaving a balance of $1,080.
SCHEDULE OF LOANS
| | |
Year ended | |
| | |
December 31,
2025 | |
| Balance, beginning of period | |
$ | 2,258 | |
| Amortized discount | |
| 239 | |
| Payments on note | |
| (1,417 | ) |
| Balance, end of period | |
$ | 1,080 | |
April
2025 Promissory Note and Guaranty
On
April 11, 2025, ENvue issued a promissory note (the “April Note”) to Alpha (the “Lender”) in the principal amount
of $360 (the “April Note Principal Amount”), together with all accrued interest thereon. The April Note has a maturity date
of June 11, 2025 (the “April Note Maturity Date”) and on the April Note Maturity Date, the aggregate unpaid April Note Principal
Amount, all accrued and unpaid interest and all other amounts payable under the April Note shall be due and payable. The April Note bears
interest at an annual rate equal to 8.0% and is payable “in kind” by adding such accrued interest to the April Note Principal
Amount.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Pursuant
to the terms of the April Note, commencing on the date of the issuance and sale of any shares common stock or Common Stock Equivalents
(as defined in the April Note) by the Company, the Lender may require ENvue to redeem all or a portion of the April Note with the proceeds of
such issuance and sale (the “Redemption Right”). The Redemption Right may be redeemed at any time after date of such issuance
and sale by the Lender providing to ENvue written notice specifying the principal amount of the Note to be redeemed (the “Redemption
Amount”), and the Redemption Amount shall be due and payable by ENvue on the second business day after the date of such notice.
The
April Note additionally provides for certain customary events of default which upon occurrence, the Lender may, at its option, declare
the entire April Note Principal Amount together with all accrued interest and all other amounts payable under the April Note immediately
due and payable, provided however, that if a bankruptcy event occurs, the April Note Principal Amount and accrued interest on the April
Note shall become immediately due and payable without any notice, declaration or other act on the part of the Lender.
In
connection with ENvue’s issuance of the April Note, on April 11, 2025, we entered into that certain Guaranty (the “Guaranty”)
in favor of the Lender, pursuant to which we have agreed to guarantee to the Lender the payment of all obligations and liabilities of
ENvue under the April Note, including, without limitation, for principal, interest and any other amounts due and payable by ENvue under
the April Note (the “Guaranteed Obligations”). Upon the occurrence of an Event of Default (as defined in the April Note),
the Guaranteed Obligations shall be deemed immediately due and payable at the election of Lender and we shall pay on demand the Guaranteed
Obligations to Lender.
On
May 19, 2025, the Company paid the April Note in full with proceeds from the 2025 Underwritten Offering, pursuant to the terms of the
April Note.
NOTE
15 - LOSS PER SHARE APPLICABLE TO COMMON STOCKHOLDERS
Basic
net loss per common share (“Basic EPS”) is computed by dividing net loss available to common stockholders by the weighted
average number of common shares outstanding during the period, including pre-funded warrants. All outstanding stock options and warrants for the year ended December
31, 2025, and 2024 have been excluded from the calculation of the diluted net loss per share because all such securities are anti-dilutive
for all periods presented.
The
following table sets forth the computation of the net loss per share for the period presented:
SCHEDULE OF NET LOSS PER SHARE
| | |
| | |
| |
| | |
December 31 | |
| | |
2025 | | |
2024 | |
| Numerator: | |
$ | | | |
$ | | |
| Net loss | |
| (18,185 | ) | |
| (3,705 | ) |
| Dividend on Convertible Preferred Series X | |
| (1,700 | ) | |
| - | |
| Dividend on Convertible Preferred Series H | |
| (396 | ) | |
| - | |
| Deemed contribution (See note 11) | |
| (399 | ) | |
| - | |
| Deemed contribution on repurchase of Preferred
Series X | |
| 90 | | |
| - | |
| Deemed contribution on
modification of Preferred Series X | |
| 3,815 | | |
| - | |
| Net
loss available to common stockholders | |
$ | (16,775 | ) | |
$ | (3,705 | ) |
The
following table summarizes the Company’s securities, in common stock equivalents, which have been excluded from the calculation
of dilutive loss per share as their effect would be anti-dilutive:
[*]
SUMMARY OF COMMON SHARE EQUIVALENTS BEEN EXCLUDED FROM DILUTIVE LOSS PER SHARE AS ANTI-DILUTIVE
| | |
| | |
| |
| | |
December 31 | |
| | |
2025 | | |
2024 | |
| Stock options - employee and non-employee | |
$ | 4,411 | | |
$ | 4,506 | |
| Warrants | |
| 1,145,585 | | |
| 29,011 | |
| Total | |
$ | 1,149,996 | | |
$ | 33,517 | |
The
diluted loss per share equals basic loss per share for year ended December 31, 2025, and 2024 because the Company had a net loss and
the impact of the assumed exercise of stock options and the vesting of restricted stock would have been anti-dilutive.
[*] Adjusted to reflect the reverse stock
splits (see note 11).
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
NOTE
16 – SEGMENT INFORMATION
Operating
segments are defined as components of an entity for which separate financial information is available and that is regularly provided
to the Chief Operating Decision Maker (“CODM”) in deciding how to allocate resources to an individual segment and in assessing
performance. The Company’s Chief Executive Officer is the Company’s CODM. The CODM reviews financial information presented
by operating segment in making operating decisions, allocating resources, and evaluating financial performance.
The
Company conducted the business through two primary operating segments: NanoVibronix and ENvue. NanoVibronix derives revenues from selling
its products directly to patients as well as through distributor agreements. ENvue derives revenues from selling its Systems and Nasoenteral
tubes. Non-allocated administrative and other expenses are reflected in Corporate.
The
Company’s chief executive officer is its chief operating decision maker (CODM), who allocates resources to and assesses the performance
of each operating segment using information about the operating segment’s loss from operations.
SCHEDULE
OF SEGMENT INFORMATION
Goodwill
and Assets
| | |
NanoVibronix | | |
ENvue | | |
Corporate | | |
Total | |
| Balance sheet
at December 31, 2025 | |
| | | |
| | | |
| | | |
| | |
| Goodwill | |
$ | - | | |
$ | 29,082 | | |
$ | - | | |
$ | 29,082 | |
| Assets | |
$ | 1,924 | | |
$ | 39,200 | | |
$ | - | | |
$ | 41,124 | |
| | |
| | | |
| | | |
| | | |
| | |
| Balance sheet at December 31, 2024 | |
| | | |
| | | |
| | | |
| | |
| Goodwill | |
$ | - | | |
$ | - | | |
$ | - | | |
$ | | |
| Assets | |
$ | 3,629 | | |
$ | - | | |
$ | - | | |
$ | 3,629 | |
Segment
operating results
| Year ended
December 31, 2025: | |
NanoVibronix | | |
ENvue | | |
Corporate | | |
Total | |
| Revenues | |
$ | 1,857 | | |
$ | 696 | | |
$ | - | | |
$ | 2,553 | |
| Cost of revenues | |
| 1,370 | | |
| 1,030 | | |
| - | | |
| 2,400 | |
| Research and development | |
| 1,088 | | |
| 674 | | |
| - | | |
| 1,762 | |
| Selling and marketing | |
| 541 | | |
| 1,952 | | |
| - | | |
| 2,493 | |
| General and administrative | |
| 2,102 | | |
| 2,337 | | |
| 3,194 | | |
| 7,633 | |
| Impairment expense | |
| - | | |
| 11,154 | | |
| - | | |
| 11,154 | |
| Total operating loss | |
$ | (3,244 | ) | |
$ | (16,451 | ) | |
$ | 3,194 | | |
$ | (22,889 | ) |
| Year ended
December 31, 2024: | |
NanoVibronix | | |
ENvue | | |
Corporate | | |
Total | |
| Revenues | |
$ | 2,558 | | |
$ | - | | |
$ | - | | |
$ | 2,558 | |
| Cost of revenues | |
| 1,050 | | |
| - | | |
| - | | |
| 1,050 | |
| Research and development | |
| 909 | | |
| - | | |
| - | | |
| 909 | |
| Selling and marketing | |
| 720 | | |
| - | | |
| - | | |
| 720 | |
| General and administrative | |
| 3,461 | | |
| - | | |
| - | | |
| 3,461 | |
| Total operating loss | |
$ | (3,582 | ) | |
$ | - | | |
$ | - | | |
$ | (3,582 | ) |
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Geographic
Information and Major Customer Data
The
following is a summary of revenues within geographic areas:
SUMMARY
OF REVENUE WITHIN GEOGRAPHIC AREAS
| | |
2025 | | |
2024 | |
| | |
Year
ended December 31 | |
| | |
2025 | | |
2024 | |
| United States | |
$ | 2,510 | | |
$ | 2,450 | |
| Europe | |
| 8 | | |
| 17 | |
| Australia/New Zealand | |
| 20 | | |
| 41 | |
| Other | |
| 15 | | |
| 50 | |
| Total | |
$ | 2,553 | | |
$ | 2,558 | |
Major Customer Data as a Percentage of Total Revenues
The following is a summary of revenues:
SCHEDULE
OF PERCENTAGE OF TOTAL REVENUES
| | |
2025 | | |
2024 | |
| | |
Year ended December 31 | |
| | |
2025 | | |
2024 | |
| | |
| | |
| |
| Customer A | |
| 31 | % | |
| 36 | % |
| Customer B | |
| 23 | % | |
| 19 | % |
| Customer C | |
| 14 | % | |
| 34 | % |
| Total | |
| 68 | % | |
| 88 | % |
NOTE
17 – FAIR VALUE
Financial
Liabilities Measured at Fair Value on a Recurring Basis
There
were no transfers between Level 3 during the year ended December 31, 2025, and 2024.
The
following table presents changes in Level 3 asset and liability measured at fair value for the year ended December 31, 2025 and 2024:
SCHEDULE
OF CHANGES IN LEVEL 3 AND LIABILITY MEASURED AT FAIR VALUE
| | |
Warrants
Liability | |
| Balance – December 31, 2024 | |
$ | - | |
| Issuance
– warrant liability | |
| 8,078 | |
| Reclassification of warrant liability to equity | |
| (1,067 | ) |
| Fair
Value adjustments – warrant liability | |
| (6,204 | ) |
| Balance – December 31, 2025 | |
$ | 807 | |
The
following table sets forth the Company’s assets and liabilities which are measured at fair value on a recurring basis by level
within the fair value hierarchy:
SCHEDULE OF ASSETS AND LIABILITIES MEASURED AT FAIR VALUE
| | |
Level
I | | |
Level
II | | |
Level
III | | |
Total | |
| | |
Fair
Value Measurements as of December 31, 2025 | |
| | |
Level
I | | |
Level
II | | |
Level
III | | |
Total | |
| Warrant liability | |
$ | - | | |
| - | | |
| 807 | | |
| 807 | |
NOTE
18 - COMMITMENTS AND CONTINGENCIES
Pending
and settled litigation
On
February 26, 2021, Protrade Systems, Inc. (“Protrade”) filed a Request for Arbitration (the “Request”) with the
International Court of Arbitration (the “ICA”) of the International Chamber of Commerce alleging the Company is in breach
of an Exclusive Distribution Agreement dated March 7, 2019 (the “Exclusive Distribution Agreement”) between Protrade and
the Company. Protrade alleges, in part, that the Company has breached the Exclusive Distribution Agreement by discontinuing the manufacture
of the DV0057 Painshield MD device in favor of an updated 10-100-001 Painshield MD device. Protrade claims damages estimated at $3 million.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
On
March 15, 2022, the arbitrator issued a final award which determined that (i) the Company had the right to terminate the Exclusive Distribution
Agreement; (ii) the Company did not breach the duty of good faith and fair dealing with regard to the Exclusive Distribution Agreement;
and (iii) the Company did not breach any confidentiality obligations to Protrade. Nevertheless, the arbitrator determined that the Company
did not comply with the obligation to supply Protrade with a year’s supply of patches, and awarded Protrade $1,500, which consists
of $1,432 for “lost profits” and $68 as reimbursement of arbitration costs, on the grounds that the Company allegedly failed
to supply Protrade with certain patches utilized by users of DV0057 Painshield MD device. The arbitrator based the decision on the testimony
of Protrade’s president who asserted that a user would use in excess of 33 patches per each device. The Company believes that the
number of patches per device alleged by Protrade is grossly inflated, and that these claims were not properly raised before the arbitrator.
Accordingly, on April 13, 2022, the Company submitted an application for the correction of the award which the arbitrator denied on June
22, 2022.
On
July 22, 2022, the Company filed a cross-motion seeking to vacate arbitration award on the grounds that the arbitrator exceeded her authority,
that the award was procured by fraud, and that the arbitrator failed to follow procedures established by New York law. In particular,
the Company averred in its motion that Protrade’s witness made false statements in arbitration, and that the arbitrator resolved
a claim that was never raised by Protrade and that has no factual basis.
On
October 3, 2022, the court issued a decision granting Protrade its petition to confirm the award and denying the cross-motion.
On
November 9, 2022, the Company filed a motion to re-argue and renew its cross-motion to vacate the arbitration decision based on newer
information that was not available during the initial hearing. On the same day, the Company also filed a notice of appeal with the Appellate
Division, Second Department. On March 21, 2023, the court denied the motion to re-argue and renew.
On
July 10, 2023, the Company filed its appeal with the Appellate Division, Second Department. That appeal is now fully briefed. In February
2025, the Second Department informed counsel for the Company that the Second Department was beginning to process the appeal for calendaring
with oral arguments to start by the end of May 2025.
On
March 30, 2026, the Appellate Division of the Second Department issued a decision and order which affirmed the judgment of the
Supreme Court, Nassau County in its entirety and dismissed the appeal, stating inter alia that “NanoVibronix failed to
establish that the arbitration award violated a strong public policy, was irrational, procured by fraud, or clearly exceeded a
specifically enumerated limitation of the arbitrator’s power”. It further held that NanoVibronix had not demonstrated
that it had shown sufficient new facts to the court on the motion to renew. The decision was conclusory and did not analyze the
facts as argued by NanoVibronix nor did it distinguish them specifically. The Company is currently 24.1 reviewing the decision and
considering its available alternatives.
As
of December 31, 2025, the Company accrued the amount of the arbitration award to Protrade of approximately $2,252, including interest which is classified in “Other accounts payable and accrued expenses”.
NOTE
19 – RELATED PARTY TRANSACTION
Aurora
Cassirer served as a member of the Company’s board of directors until her resignation effective April 1, 2025. During her tenure,
Ms. Cassirer also served on the audit committee, the corporate governance and nominating committee, and the compensation committee. Her
resignation was not in connection with any disagreement with the Company on any matter relating to the Company’s operations, policies,
or practices, or any other matter. Ms. Cassirer is an attorney but did not provide any legal services or legal advice to the Company
during the period that she was a member of the board.
The
firm FisherBroyles LLP handled all of the Company’s Protrade litigation and appeals through December 31, 2024. For the year ended
December 31, 2024, the Company was not billed and did not pay any legal fees to FisherBroyles. On January 1, 2024, Ms. Cassirer and the
lawyers responsible for handling the Company’s Protrade litigation left FisherBroyles to join the firm Pierson Ferdinand LLP, which
since that date has currently been the sole firm handling all of the Company’s Protrade litigation and appeals. For the year ended
December 31, 2025 and 2024, Pierson Ferdinand was paid $323 and $69, respectively, while Ms. Cassirer was a member of the board. As in
prior years, Ms. Cassirer has not provided any legal services or legal advice to the Company.
On
January 17, 2025, ENvue issued a Consolidated Secured Note (as amended, the “Alpha Note”) in the aggregate principal
amount of $2,497,
to Alpha Capital Anstalt (“Alpha”), see note 14. Prior to the ENvue Merger, Alpha was the principal shareholder of
ENvue, and following the completion of the ENvue Merger, Alpha continues to be a shareholder of the Company, however subsequent to
the merger they are subject to a 4.99% beneficial ownership limitation. As of December 31, 2025, Alpha also holds 40,996
of Series X Preferred Shares (see Note 4).
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
On February 13, 2025, the Company entered into a Securities Purchase Agreement (the “PIPE Purchase Agreement”) with Alpha, pursuant
to which the Company sold in a private placement senior convertible debenture (the “Debenture”) due the earlier of (i) the
date that is the 30-day anniversary of the effective date of stockholder approval of the issuance of the shares of common stock upon
the conversion of the debentures and (ii) November 13, 2025 (the date that is nine months following the date of issuance of the Debenture)
(“Maturity Date”), having an aggregate principal amount (the “Principal Amount”) of $
500
(the “Debenture Transaction”). The closing of the Debenture Transaction occurred on February 14, 2025—see note 13. As of December 31, 2025, no amounts are outstanding under this debenture.
On
February 14, 2025, pursuant to the terms of the Merger Agreement and in connection with the Merger and the appointment of Doron Besser
and Zeev Rotstein (together, the “Indemnitees”) to the Board of Directors, the Company and each of the Indemnitees entered
into the Company’s standard form of indemnification agreement, which such indemnification agreements provide that the Company shall
indemnify the Indemnitees to the fullest extent of permitted by applicable law in effect on the date hereof or as amended to increase
the scope of permitted indemnification, against expenses, losses, liabilities, judgments, fines, penalties and amounts paid in settlement
(including all interest, taxes, assessments and other charges in connection therewith) incurred by Indemnitee or on Indemnitee’s
behalf in connection with any proceeding in any way connected with, resulting from or relating to Indemnitee’s corporate status.
On
April 11, 2025, ENvue issued a promissory note (the “April Note”) to Alpha in the principal amount of $360 (the “April
Note Principal Amount”), together with all accrued interest thereon. The April Note has a maturity date of June 11, 2025 (the “April
Note Maturity Date”) and on the April Note Maturity Date, the aggregate unpaid April Note Principal Amount, all accrued and unpaid
interest and all other amounts payable under the April Note shall be due and payable. The April Note bears interest at an annual rate
equal to 8.0% and is payable “in kind” by adding such accrued interest to the April Note Principal Amount—see note
14.
On
July 18, 2025, Company entered into a Securities Purchase Agreement (the “Purchase Agreement”) with Alpha, pursuant to which it agreed to sell to them (i) an aggregate of 8,889
shares of the Company’s newly-designated Series H Convertible Preferred Stock, with a par value of $0.001 per share and a stated
value of $1 per share (the “Stated Value”), initially convertible into up to 880,099 shares of the Company’s
common stock, par value $0.001 per share (the “Common Stock”) at an initial conversion price of $10.10 per share (the
“Preferred Stock”) and (ii) warrants to acquire up to an aggregate of 467,836 shares of Common Stock (the “Warrants”)
at an exercise price of $22.50 (the “Private Placement” and such closing, the “Initial Closing”).
Pursuant
to the terms of the Purchase Agreement, the Company has also agreed to issue 2,222 shares of Preferred Stock with a total stated value
of $2,222 in a second closing, subject to the satisfaction of customary closing conditions—see note 11.
On
September 16, 2025, the Company entered into a securities purchase agreement with Alpha, pursuant to which
the Company agreed to issue and sell (i) 74,114 shares of the Company’s Common Stock, and (ii) prefunded warrants to purchase up
to 217,090 shares of Common Stock in a registered direct offering for $2.04 million, pursuant to an effective shelf registration statement
on Form S-3. The offering price was $7.01 per share of Common Stock and $7.009 per Prefunded Warrant, which is the price of each share
of Common Stock sold in the Offering, minus the $0.001 exercise price per Prefunded Warrant. The net proceeds from the Offering were
approximately $1.88 million, after deducting placement agent fees of $163—see note 11.
NOTE
20 – INCOME TAXES
U.S. tax
reform:
On
December 22, 2017, the U.S. government enacted the Tax Cuts and Jobs Act (the “Tax Act”). The Tax Act includes significant
changes to the U.S. corporate income tax system including but not limited to: a federal corporate rate reduction from 35% to 21%; creation
of the base erosion anti-abuse tax (“BEAT”), introduction of the Global Intangible Low Taxed Income (“GILTI”)
provisions; the transition of U.S. international taxation from a worldwide tax system to a modified territorial tax system; modifications
to the allowance of net business interest expense deductions; modification of net operating loss provisions; changes to 162(m) limitation
rules and bonus depreciation provisions. The change to a modified territorial tax system resulted in a one-time U.S. tax liability on
those earnings which have not previously been repatriated to the U.S. (the “Transition Tax”), with future dividend distributions
not subject to U.S. federal income tax when repatriated. A majority of the provisions in the Tax Act became effective January 1, 2018.
The
Tax Act added a new code section 951A, which requires a U.S. shareholder of a Controlled Foreign Corporation (“CFC”) to include
in current taxable income, its GILTI in a manner similar to Subpart F income. The statutory language also allows a deduction for corporate
shareholders equal to 50% of the GILTI inclusion, which would be reduced to 37.5% starting in 2026. In general, GILTI imposes a tax on
the net income of foreign corporate subsidiaries in excess of a deemed return on their tangible assets. The Company is subject to GILTI
for 2018 and future periods. The Company is electing to account for the income tax effects of GILTI as a “period cost,” an
income tax expenses in the year the tax is incurred.
On
July 4, 2025, the U.S. government enacted the One Big Beautiful Bill Act (“OBBBA”). For the year ending December 31, 2025,
the OBBBA the law brought a return to 100% bonus depreciation, full expensing of domestic R&E costs, and modified Section 163(j)
rules previously enacted by the TCJA. The law also brought changes related to the GILTI regime, which will now be called Net CFC Tested
Income, a reduction of the Section 250 deduction from 50% to 40%, and the increase in allowable foreign tax credits. The non-domestic
changes generally result in a slight increase in effective tax rate on certain foreign income, and take place for years ending after
December 31, 2025. Ceva has implemented the domestic changes applicable to the year ending December 31, 2025, and will continue to analyze
the impact on foreign changes for the year ending December 31, 2026. The adoption of the OBBBA did not have a material impact on the
Company’s consolidated financial statements or related deferred tax balances.
Foreign
tax:
Tax
rates applicable to the income of the Israeli subsidiaries:
The
Israeli corporate tax rate in 2025 and 2024 is 23%. One of the Company’s Israeli subsidiaries has final tax assessments through 2017.
Tax
loss carryforwards:
As
of December 31, 2025, the Company and its subsidiary had federal and state net operating loss carryforwards for income tax purposes of
approximately $55,320
and $6,004,
respectively. Of the federal net operating loss carryforward, approximately $41,362
may be carried forward indefinitely. The remaining approximately
$13,959 of
federal net operating loss carryforward is subject to a 20-year
carryforward limitation but may be used to fully offset taxable income in the year of utilization. The state net operating loss carryforwards
are subject to varying carryforward periods under applicable state tax laws.
Utilization
of the U.S. federal and state net operating loss carryforwards may be subject to annual limitations under Section 382 of the
Internal Revenue Code of 1986, as amended, in the event of a cumulative change in ownership of more than 50% within any three-year
period. The Company has not performed a Section 382 analysis as of the balance sheet date; however, management does not believe that
any such limitation would have a material impact on the financial statements, as the related deferred tax assets are fully offset by a
valuation allowance.
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Tax
returns:
Income tax returns are filed in the United States
and various state jurisdictions. The Company is not currently under examination by income tax authorities in federal or state jurisdictions.
Due to net operating loss carryforwards, the Company’s returns remain open for all prior years.
Income
tax expense is comprised of the following:
SCHEDULE
OF PROVISION FOR INCOME TAXES EXPENSES
| | |
| 1 | | |
| 2 | |
| | |
Year ended December 31, | |
| | |
2025 | | |
2024 | |
| Current Tax | |
| | | |
| | |
| Federal | |
$ | - | | |
$ | - | |
| State | |
| 1 | | |
| - | |
| Foreign | |
| 65 | | |
| 19 | |
| Total | |
$ | 66 | | |
$ | 19 | |
| | |
| | | |
| | |
| Deferred Tax | |
| | | |
| | |
| Federal | |
$ | (331 | ) | |
$ | - | |
| State | |
| (42 | ) | |
| - | |
| Foreign | |
$ | - | | |
| - | |
| Total | |
$ | (373 | ) | |
$ | - | |
| | |
| | | |
| | |
| Total taxes on income (benefit) | |
$ | (307 | ) | |
$ | 19 | |
Loss
before taxes:
SCHEDULE
OF INCOME BEFORE TAXES ON DOMESTIC AND FOREIGN
| Foreign | |
| (221 | ) | |
| (89 | ) |
| | |
Year ended December 31, | |
| | |
2025 | | |
2024 | |
| | |
| | |
| |
| Domestic | |
$ | 18,713 | | |
$ | 3,775 | |
| Foreign | |
| (221 | ) | |
| (89 | ) |
Reconciliation
between the Company’s effective tax rate and the U.S. statutory rate:
SCHEDULE
OF EFFECTIVE INCOME TAX RATE RECONCILIATION
The
company adopted ASU 2023-09 for the year ended December 31, 2025, on a prospective basis. A reconciliation between the theoretical
tax expense, assuming all income is taxed at the statutory tax rate applicable to income of the Company, and the actual tax expense as
reported in the consolidated statements of loss is as follows:
| Nontaxable or nondeductible items: | |
| | | |
| | |
| | |
Year ended December 31, | |
| | |
2025 | |
| | |
Amount | | |
Rate | |
| US federal statutory tax rate | |
$ | (3,883 | ) | |
| 21 | % |
| State and local income taxes, net of federal | |
| (42 | ) | |
| 0.23 | % |
| Foreign tax effects | |
| 32 | | |
| (0.17 | )% |
| Nontaxable or nondeductible items: | |
| | | |
| | |
| Goodwill impairment | |
| 2,207 | | |
| (11.93 | )% |
| Warrants remeasurement | |
| (1,303 | ) | |
| 7.05 | % |
| Other | |
| 152 | | |
| (0.82 | )% |
| Change in valuation allowance | |
| 2,444 | | |
| (13.22 | )% |
| Other adjustments | |
| 86 | | |
| (0.47 | )% |
| Income tax expense (benefit) | |
$ | (307 | ) | |
| 1.66 | % |
Reconciliation
between the Company’s effective tax rate and the U.S. statutory rate prior to the adoption of ASU 2023-09:
| | |
Year ended December 31, | |
| | |
2024 | |
| Federal income tax benefit at statutory rate | |
| 21.00 | % |
| State income taxes, net of federal benefit | |
| 0.31 | % |
| Foreign rate differential | |
| 0.04 | % |
| Permanent Items | |
| (0.04 | )% |
| Change in valuation allowance | |
| (21.51 | )% |
| Return to provision adjustments | |
| 0.40 | % |
| Other | |
| 0.30 | % |
| Effective tax rate | |
| 0.51 | % |
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
Deferred
income taxes:
Deferred
income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial
purposes and the amounts used for income tax purposes. Significant components of the Company’s deferred tax assets are as follows:
SCHEDULE
OF DEFERRED TAX ASSETS
| |
|
2024 |
|
|
2023 |
|
| |
|
Year ended December 31, |
|
| |
|
2025 |
|
|
2024 |
|
| Deferred tax assets: |
|
|
|
|
|
|
|
|
| Net operating loss carryforward |
|
$ |
12,778 |
|
|
$ |
9,032 |
|
| R&D expense |
|
|
164 |
|
|
|
207 |
|
| Capital loss carryforward |
|
|
5 |
|
|
|
6 |
|
| Arbitration and other accrued expenses |
|
|
379 |
|
|
|
385 |
|
| Stock compensation |
|
|
539 |
|
|
|
581 |
|
| Lease liability |
|
|
29 |
|
|
|
17 |
|
| Other |
|
|
42 |
|
|
|
- |
|
| Total deferred tax assets before valuation allowance |
|
|
13,936 |
|
|
|
10,228 |
|
| Valuation allowance |
|
|
(13,009 |
) |
|
|
(10,204 |
) |
Net deferred tax asset |
|
|
927 |
|
|
|
24 |
|
| Deferred tax liabilities: |
|
|
|
|
|
|
|
|
| Property and equipment |
|
|
(15 |
) |
|
|
(5 |
) |
| Intangible Assets |
|
|
(885 |
) |
|
|
- |
|
| Right-of-use Asset |
|
|
(27 |
) |
|
|
(19 |
) |
| Net deferred tax asset |
|
$ |
- |
|
|
$ |
- |
|
The net change in the total valuation allowance for
the year ended December 31, 2025 was a decrease of $2,805. In assessing the realization of deferred tax assets, management considers whether
it is more likely than not that some or all of the deferred tax assets will not be realized. The ultimate realization of deferred
tax assets depends on the generation of future taxable income during the periods in which those temporary differences and tax loss carryforward
are deductible. Management considers the projected taxable income and tax-planning strategies in making this assessment.
The Company recorded a valuation allowance for certain
of its deferred tax assets. The Company concluded that, based on the weight of available positive and negative evidence, it was more likely
than not that the deferred tax assets would not be recoverable due to uncertainty regarding future taxable income. In assessing the realizability
of deferred tax assets, the key assumptions used to determine positive and negative evidence included the Company’s current trends
related to actual taxable earnings or losses, and expected future reversals of existing taxable temporary differences, as well as projections
for future annual results.
The Company considers the earnings of its non-U.S.
subsidiary to be indefinitely invested outside the United States on the basis of estimates that future domestic cash generation will be
sufficient to meet future domestic cash needs and our specific plans for reinvestment of those subsidiary earnings. If the Company does
decide to repatriate the foreign earnings, we would need to adjust our income tax provision in the period we determined that the earnings
will no longer be indefinitely invested outside the United States.
Income tax payments:
Pursuant to the disclosure requirements of ASU 2023-09,
below is a summary of income taxes paid, net of refunds received, by jurisdiction for the year ended December 31,2025:
SUMMARY
OF INCOME TAXES PAID, NET OF REFUNDS RECEIVED
| | |
Year ended December 31, | |
| | |
2025 | |
| Cash taxes paid (refunds received) | |
| | |
| Federal income taxes | |
| 32 | |
| Foreign: | |
| | |
| Israel | |
| 20 | |
| Income taxes, net of amounts refunded | |
$ | 52 | |
ENVUE
MEDICAL, INC.
Notes
to Consolidated Financial Statements
(Amounts
in thousands except share and per share data)
NOTE 21 – IMPAIRMENT OF GOODWILL AND LONG-LIVED ASSETS
During
the year ended December 31, 2025, the Company performed its annual goodwill impairment test for the ENvue reporting unit. In addition, based on the presence of indicators of impairment identified during the period, the Company also evaluated the recoverability of its long-lived assets. As a result of these assessments,
the Company recognized impairment charges related to goodwill and certain intangible assets.
Long-lived
asset impairment
The
Company evaluated the long-lived assets included in the ENvue reporting unit under ASC 360, Property, Plant, and Equipment
(“ASC 360”). Long-lived assets were tested for impairment at the asset group level. The Company identified the asset
group as the entire ENvue reporting unit, which represents the lowest level for which identifiable cash flows are largely
independent of the cash flows of other groups of assets and liabilities.
As the ENvue asset
group did not pass the step one recoverability test on an undiscounted cash flow basis, the Company then compared the fair value of the
asset group to its carrying value. Accordingly to estimate the fair value of the asset group, the Company utilized an income-based valuation
approach by means of a discounted cash flow method, based on market participant assumptions. The assumptions used to estimate the fair
value using a discounted cash flow method included forecasted revenue and EBITDA and long-term expectations for growth rates
and operating profit margin, expected needs for annual capital expenditure and net working capital, and a market-participant discount
rate derived via the capital asset pricing model. The excess of the carrying value of the asset group over the fair value
was first allocated amongst the long-lived assets (excluding goodwill), noting each individual long-lived asset within the group
may not be impaired below its individual fair value.
As
a result, the Company recognized impairment charges of $232 related to proprietary technology, $32 related to tradenames and trademarks,
and $381 related to customer relationships. No other long-lived assets were impaired, as their carrying values did not exceed their respective
fair values.
Goodwill
impairment
Following
the impairment assessment of long-lived assets, the Company performed its annual goodwill impairment test for the ENvue reporting unit.
The fair value of the reporting unit was estimated using a discounted cash flow methodology consistent with that described above and
was categorized as a Level 3 measurement within the fair value hierarchy due to the use of significant unobservable inputs.
As
of December 31, 2025, the carrying value of the ENvue reporting unit exceeded its estimated fair value. Accordingly, the Company recognized
a non-cash goodwill impairment charge of $10,509 for the year ended December 31, 2025, which is included in operating expenses.
Total
Impairment
Accordingly,
for the year ended December 31, 2025, the Company recognized a total impairment charge of $11,154 related to the ENvue reporting unit.
Future
changes in projected operating results, including those driven by economic conditions or operational factors, could result in additional
impairment charges, which may be material.
NOTE
22 - SUBSEQUENT EVENTS
Certificate of Amendment to Series H Preferred Stock
On January 30, 2026, the Company entered into the Series H Amendment Agreement with the Required Holders (as defined
in the Series H Amendment Agreement). Pursuant to the Series H Amendment Agreement, the Required Holders agreed to amend the Series H
Certificate of Designations by filing the Series H Certificate of Amendment to the Series H Certificate of Designations with the Secretary
of State of the State of Delaware to remove the Floor Price (as defined in the Series H Certificate of Designations) in consideration
of the holders of the Series H Preferred Stock exercising $2,500,000 of the Additional Investment Right (as such concept is described
in the Securities Purchase Agreement by and between the Company and the holders of the Series H Preferred Stock
Employment
Agreement Amendment – Doron Besser
On
February 2, 2026, ENvue Medical Israel, Ltd., a wholly owned subsidiary of the Company, entered into a first amendment to Dr. Doron
Besser’s Amended and Restated Employment Agreement. The amendment revised the timing of the initial grant of restricted stock
units (“RSUs”) and provides that additional restricted stock units (“RSUs”) may be granted on a
quarterly basis, subject to Board approval, to maintain Dr. Besser’s target equity ownership of 9%
on a fully diluted basis. The amendment also provides for a prorated equity true-up in the event of certain qualifying
terminations. The initial grant was subject to the approval of the Compensation Committee, which was approved on April 4,
2026.
Chairman Agreement – David Johnson
On February 2, 2026, the Company entered into a Chairman Agreement with David Johnson, pursuant to which he will serve as Chairman of
the Board. Under the agreement, Mr. Johnson will receive compensation of $10,000 per month, reimbursement of reasonable expenses, and
an equity award representing approximately 3.5% of the Company’s fully diluted common stock, subject to vesting. The agreement has
an initial one-year term with automatic renewals, unless terminated upon notice by either party.
Loan Amendment
On March 25, 2026, the Company and Alpha entered into an amendment to a loan originally
issued on January 17, 2025, in the principal amount of approximately $2.5 million, as previously amended on February 13, 2025. The amendment
modified the maturity date, extending it to December 31, 2026. All other terms of the loan remain unchanged.
Index
to Exhibits
| Exhibit
No. |
|
Description |
| |
|
|
| 1.1 |
|
Underwriting Agreement, by and between the Company and Dawson James Securities, Inc., dated as of May 14, 2025 (incorporated by reference to Exhibit 1.1 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 2.1# |
|
Agreement
and Plan of Merger, dated February 14, 2025, by and among NanoVibronix, Inc., NVEH Merger Sub I, Inc., NVEH Merger Sub II, LLC and
ENvue Medical Holdings, Corp. (incorporated by reference to Exhibit 2.1 to the Current Report on Form 8-K filed with the Securities
and Exchange Commission on February 14, 2025). |
| |
|
|
| 3.1 |
|
Amended
and Restated Certificate of Incorporation (as presently in effect) (incorporated by reference to Exhibit 3.1 to the Current Report
on Form 8-K filed with the Securities and Exchange Commission on April 17, 2015). |
| |
|
|
| 3.2 |
|
Amended
and Restated Bylaws (incorporated by reference to Exhibit 3.2 to Amendment No. 3 to the Registration Statement on Form S-1 filed
with the Securities and Exchange Commission on April 30, 2014). |
| |
|
|
| 3.3 |
|
Certificate
of Amendment of Certificate of Incorporation (creating the Series C Preferred Stock) (incorporated by reference to Exhibit 3.3 to
Amendment No. 3 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on April 30, 2014). |
| |
|
|
| 3.4 |
|
Certificate
of Designation of Preferences, Rights and Limitations of Series D Convertible Preferred Stock (incorporated by reference to Exhibit
3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 7, 2017). |
| |
|
|
| 3.5 |
|
Certificate
of Designation, Preferences, Rights and Limitations of Series E Preferred Stock (incorporated by reference to Exhibit 4.1 to the
Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 19, 2019). |
| |
|
|
| 3.6 |
|
Certificate
of Amendment of the Amended and Restated Certificate of Designation (incorporated herein by reference to Exhibit 3.1 to the Current
Report on Form 8-K filed with the Securities and Exchange Commission on November 21, 2019). |
| |
|
|
| 3.7 |
|
Certificate
of Amendment of Certificate of Incorporation (incorporated by reference to Exhibit 3.7 to the Quarterly Report on Form 10-Q filed
with the Securities and Exchange Commission on November 15, 2021). |
| |
|
|
| 3.8 |
|
Amendment to the Amended and Restated Bylaws (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 3, 2021). |
| |
|
|
| 3.9 |
|
Certificate of Designation, Preferences, Rights and Limitations of Series F Preferred Stock (incorporated by reference to Exhibit 3.1 to the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 14, 2022). |
| |
|
|
| 3.10 |
|
Certificate of Amendment of Certificate of Incorporation (incorporated by reference to Exhibit 3.1 to the Current Report filed with the Securities and Exchange Commission on February 8, 2023). |
| |
|
|
| 3.11 |
|
Certificate
of Designations of Preferences, Rights and Limitations of Series X Non-Voting Convertible Preferred Stock, dated February 14, 2025
(incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on
February 14, 2025). |
| |
|
|
| 3.12 |
|
Certificate
of Amendment to the Amended and Restated Certificate of Incorporation, as amended, of NanoVibronix, Inc. (incorporated by reference
to Exhibit 3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on March 12, 2025). |
| |
|
|
| 3.13 |
|
Certificate of Amendment to Certificate of Designation of Preferences, Rights and Limitations of Series X Non-Voting Convertible Preferred Stock (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 3.14 |
|
Certificate of Designation of the Preferences, Rights and Limitations of Series G Convertible Preferred Stock (incorporated by reference to Exhibit 3.2 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 3.15 |
|
Certificate of Correction to Certificate of Designation of the Preferences, Rights and Limitations of Series G Convertible Preferred Stock, dated July 8, 2025 (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed on July 9, 2025). |
| |
|
|
| 3.16 |
|
Certificate of Designation of the Preferences, Rights and Limitations of Series H Convertible Preferred Stock, filed July 18, 2025 (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed on July 22, 2025). |
| |
|
|
| 3.17 |
|
Certificate of Amendment of Certificate of Incorporation of ENvue Medical, Inc. (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 12, 2025). |
| |
|
|
| 3.18 |
|
Certificate of Amendment to Certificate of Designation of Preferences, Rights and Limitations of Series H Convertible Preferred Stock (incorporated by reference to Exhibit 3.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on January 30, 2026). |
| |
|
|
| 4.1 |
|
Form
of Common Stock Certificate (incorporated by reference to Exhibit 4.2 to Amendment No. 1 to the Registration Statement on Form S-1
filed with the Securities and Exchange Commission on March 6, 2014). |
| 4.2 |
|
Form
of May 10 and May 15, 2019 Warrants (incorporated by reference to Exhibit 4.1 to the Quarterly Report on Form 10-Q filed with the
Securities and Exchange Commission on May 20, 2019). |
| |
|
|
| 4.3 |
|
Form
of Warrant (incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission
on June 26, 2019). |
| |
|
|
| 4.4 |
|
Form
of Preferred Warrant (incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K filed with the Securities and Exchange
Commission on July 31, 2019). |
| |
|
|
| 4.5 |
|
Form
of Common Warrant (incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K filed with the Securities and Exchange
Commission on July 31, 2019). |
| |
|
|
| 4.6 |
|
Form
of Warrant Amendment (incorporated by reference to Exhibit 4.10 to the Annual Report on Form 10-K filed with the Securities and Exchange
Commission on May 20, 2020). |
| |
|
|
| 4.7 |
|
Form of Placement Agent Warrant (incorporated by reference to Exhibit 4.3 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on September 1, 2023). |
| |
|
|
| 4.8 |
|
Form of Senior Convertible Debenture, issued on February 13, 2025(incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 14, 2025). |
| |
|
|
| 4.9 |
|
Amended and Restated Senior Convertible Debenture Due the Earlier of the Trigger Date and November 13, 2025 (incorporated by reference to Exhibit 10.78 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2025). |
| |
|
|
| 4.10 |
|
Form of Promissory Note, dated as of April 11, 2025, issued by ENvue Medical Holdings LLC to Alpha Capital Anstalt (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on April 11, 2025). |
| |
|
|
| 4.11 |
|
Form of Merger Pre-Funded Warrant (incorporated by reference to Exhibit 4.20 to the Company’s Registration Statement on Form S-1/A, as filed with the SEC on April 22, 2025). |
| |
|
|
| 4.12 |
|
Form of Common Warrant issued on May 16, 2025 (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 4.13 |
|
Form of Representative’s Warrant issued on May 16, 2025 (incorporated by reference to Exhibit 4.2 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 4.14 |
|
Form of Warrant dated July 22, 2025 (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed with the SEC on July 22, 2025). |
| |
|
|
| 4.15 |
|
Form of Pre-Funded Warrant dated September 17, 2025 (incorporated by reference to Exhibit 4.1 to the Current Report on Form 8-K filed with the SEC on September 17, 2025). |
| |
|
|
| 4.16* |
|
Description of Securities. |
| |
|
|
| 10.1 |
|
Fourteenth Amended and Restated Securities Purchase Agreement, dated June 16, 2014, by and between NanoVibronix, Inc. and Globis Overseas Fund, Ltd. (incorporated by reference to Exhibit 10.9 to the Registration Statement on Form 10 filed with the Securities and Exchange Commission on February 9, 2015). |
| |
|
|
| 10.2 |
|
Fourteenth Amended and Restated Securities Purchase Agreement, dated December 11, 2014, by and between NanoVibronix, Inc. and Globis Capital Partners, L.P. (incorporated by reference to Exhibit 10.10 to the Registration Statement on Form 10 filed with the Securities and Exchange Commission on February 9, 2015). |
| 10.3 |
|
Fifteenth Amended and Restated Secured Convertible Promissory Note, dated December 11, 2014, by NanoVibronix, Inc. in favor of and Globis Overseas Fund, Ltd. (incorporated by reference to Exhibit 10.11 to the Registration Statement on Form 10 filed with the Securities and Exchange Commission on February 9, 2015). |
| |
|
|
| 10.4 |
|
Fifteenth Amended and Restated Secured Convertible Promissory Note, dated December 11, 2014, by NanoVibronix, Inc. in favor of and Globis Capital Partners, L.P. (incorporated by reference to Exhibit 10.12 to the Registration Statement on Form 10 filed with the Securities and Exchange Commission on February 9, 2015). |
| |
|
|
| 10.5 |
|
Form
of Amended and Restated 2013 and 2014 Warrant to Purchase Common Stock (incorporated by reference to Exhibit 10.13 to Amendment No.
2 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on March 25, 2014). |
| |
|
|
| 10.6+ |
|
NanoVibronix,
Inc. 2004 Global Share Option Plan (incorporated by reference to Exhibit 10.14 to Amendment No. 1 to the Registration Statement on
Form S-1 filed with the Securities and Exchange Commission on March 6, 2014). |
| |
|
|
| 10.7+ |
|
Personal
Employment Agreement, dated March 1, 2008, by and between NanoVibronix (Israel 2003) Ltd and Jona Zumeris (incorporated by reference
to Exhibit 10.15 to Amendment No. 1 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on
March 6, 2014). |
| |
|
|
| 10.8+ |
|
Form
of Indemnification Agreement between NanoVibronix, Inc. and certain of its officers and directors (incorporated by reference to Exhibit
10.16 to Amendment No. 1 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on March 6,
2014). |
| |
|
|
| 10.9 |
|
Amendment
to Subscription Agreement Convertible Promissory Notes, dated February 28, 2014, by and between NanoVibronix, Inc. and the note holders
signatory thereto (incorporated by reference to Exhibit 10.17 to Amendment No. 1 to the Registration Statement on Form S-1 filed
with the Securities and Exchange Commission on March 6, 2014). |
| |
|
|
| 10.10 |
|
Second
Amendment to Subscription Agreement Series B Convertible Preferred Stock and Warrants), dated February 28, 2014, by and between NanoVibronix,
Inc. and the holders signatory thereto (incorporated by reference to Exhibit 10.19 to Amendment No. 1 to the Registration Statement
on Form S-1 filed with the Securities and Exchange Commission on March 6, 2014). |
| |
|
|
| 10.11 |
|
Third
Amendment to Subscription Agreement Series B Convertible Preferred Stock and Warrants), dated February 28, 2014, by and between NanoVibronix,
Inc. and the holders signatory thereto (incorporated by reference to Exhibit 10.20 to Amendment No. 1 to the Registration Statement
on Form S-1 filed with the Securities and Exchange Commission on March 6, 2014). |
| |
|
|
| 10.12+ |
|
NanoVibronix,
Inc. 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.27 to Amendment No. 3 to the Registration Statement on
Form S-1 filed with the Securities and Exchange Commission on April 30, 2014). |
| |
|
|
| 10.13+ |
|
First
Amendment to Personal Employment Agreement, dated June 16, 2014, by and between NanoVibronix, Inc. and Dr. Jona Zumeris (incorporated
by reference to Exhibit 10.29 to Amendment No. 8 to the Registration Statement on Form S-1 filed with the Securities and Exchange
Commission on June 23, 2014). |
| |
|
|
| 10.14 |
|
Services
Agreement, dated March 25, 2015, by and between Multigon Industries, Inc. and NanoVibronix, Inc. (incorporated by reference to Exhibit
10.35 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.15+ |
|
Employment
Agreement, dated March 25, 2015, by and between William Stern and NanoVibronix, Inc. (incorporated by reference to Exhibit 10.36
to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.16+ |
|
Letter
Agreement, dated March 25, 2015, by and between NanoVibronix, Inc. and Martin Goldstein (incorporated by reference to Exhibit 10.39
to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| 10.17+ |
|
Form
of Incentive Stock Option Award Agreement under the 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.40 to
the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.18+ |
|
Form
of Nonqualified Stock Option Award Agreement under the 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.41
to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.19+ |
|
Form
of Restricted Stock Award Agreement under the 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.42 to the Annual
Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.20+ |
|
Form
of 3(i) Award Agreement under the Israeli Appendix to the 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.43
to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.21+ |
|
Form
of 102 Award Agreement under the Israeli Appendix to the 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.44
to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 30, 2015). |
| |
|
|
| 10.22+ |
|
Employment
Agreement, dated October 13, 2016, by and between NanoVibronix, Inc. and Brian Murphy (incorporated by reference to Exhibit 10.3
to the Current Report on Form 8-K filed with the Securities and Exchange Commission on October 19, 2016). |
| |
|
|
| 10.23 |
|
Form
of Convertible Promissory Note (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities
and Exchange Commission on March 7, 2017). |
| |
|
|
| 10.24 |
|
Convertible
Promissory Note, dated March 23, 2017, by and between NanoVibronix, Inc. and an individual investor (incorporated by reference to
Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on March 27, 2017). |
| |
|
|
| 10.25+ |
|
First
Amendment to Nonqualified Stock Option Agreement, dated March 30, 2017, between NanoVibronix, Inc. and Ira A. Greenstein (incorporated
by reference to Exhibit 10.51 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2017) |
| |
|
|
| 10.26+ |
|
First
Amendment to Nonqualified Stock Option Agreement, dated March 30, 2017, between NanoVibronix, Inc. and Ira A. Greenstein (incorporated
by reference to Exhibit 10.52 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2017). |
| |
|
|
| 10.27+ |
|
Offer
Letter, dated October 14, 2016, between NanoVibronix, Inc. and Christopher M. Fashek (incorporated by reference to Exhibit 10.1 to
the Current Report on Form 8-K filed with the Securities and Exchange Commission on October 19, 2016). |
| |
|
|
| 10.28+ |
|
Nonqualified
Stock Option Agreement, dated October 14, 2016, between NanoVibronix, Inc. and Christopher M. Fashek (incorporated by reference to
Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on October 19, 2016). |
| |
|
|
| 10.29 |
|
Form
of Convertible Promissory Note (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities
and Exchange Commission on May 5, 2017). |
| |
|
|
| 10.30 |
|
Form
of Letter Agreement, dated September 7, 2017, between NanoVibronix, Inc. and holders of the 2017 Notes (incorporated by reference
to Exhibit 10.1 to the Current Report on Form 8-K/A filed with the Securities and Exchange Commission on September 14, 2017). |
| |
|
|
| 10.31 |
|
Consulting
Agreement dated as of February 21, 2019, between NanoVibronix, Inc and Bespoke Growth Partners, Inc. (incorporated by reference to
Exhibit 10.36 to the Annual Report on Form 10-K/A filed with the Securities and Exchange Commission on May 13, 2019). |
| |
|
|
| 10.32 |
|
Convertible
Promissory Note (incorporated by reference to Exhibit 10.37 to the Annual Report on Form 10-K/A filed with the Securities and Exchange
Commission on May 13, 2019). |
| 10.33 |
|
Convertible
Promissory Note (incorporated by reference to Exhibit 10.38 to the Annual Report on Form 10-K/A filed with the Securities and Exchange
Commission on May 13, 2019). |
| |
|
|
| 10.34 |
|
Form
of Warrant (incorporated by reference to Exhibit 10.39 to the Annual Report on Form 10-K/A filed with the Securities and Exchange
Commission on May 13, 2019). |
| |
|
|
| 10.35 |
|
Convertible
Promissory Note (Globis), May 10, 2019 (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q filed with
the Securities and Exchange Commission on May 20, 2019). |
| |
|
|
| 10.36 |
|
Convertible
Promissory Note (AiGH), May 15, 2019 (incorporated by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q filed with the
Securities and Exchange Commission on May 20, 2019). |
| |
|
|
| 10.37+ |
|
CFO
Consulting Agreement, dated as of June 1, 2019, between NanoVibronix Inc. and James S. Cardwell (incorporated by reference to Exhibit
10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on June 4, 2019). |
| |
|
|
| 10.38 |
|
Securities
Purchase Agreement, dated as of June 21, 2019, by and among the Company and each investor identified on the signature pages thereto
(incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on
June 26, 2019). |
| |
|
|
| 10.39 |
|
Securities
Purchase Agreement, dated as of July 31, 2019, by and among the Company and each investor identified on the signature pages thereto
(incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on
July 31, 2019). |
| |
|
|
| 10.40 |
|
Securities
Purchase Agreement, dated as of July 31, 2019, by and among the Company and each investor identified on the signature pages thereto
(incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on
July 31, 2019). |
| |
|
|
| 10.41 |
|
Form
of Note (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission
on June 26, 2020). |
| |
|
|
| 10.42 |
|
Form
of Warrant (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission
on June 26, 2020). |
| |
|
|
| 10.43 |
|
Note
with Cross River Bank (SBA-Payroll Protection Program loan) dated May 14, 2020 (incorporated by reference to Exhibit 10.3 to the
Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 19, 2020). |
| |
|
|
| 10.44+ |
|
Employment
Agreement, dated as of October 5, 2020, between NanoVibronix, Inc. and Stephen Brown (incorporated by reference to Exhibit 10.1 to
the Current Report on Form 8-K filed with the Securities and Exchange Commission on October 8, 2020). |
| |
|
|
| 10.45+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Brian Murphy (incorporated by reference
to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| |
|
|
| 10.46+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Christopher Fashek (incorporated
by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| |
|
|
| 10.47+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Martin Goldstein (incorporated
by reference to Exhibit 10.3 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| 10.48+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Michael Ferguson (incorporated
by reference to Exhibit 10.4 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| |
|
|
| 10.49+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Stephen Brown (incorporated by
reference to Exhibit 10.5 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| |
|
|
| 10.50+ |
|
Option
Cancellation and Release Agreement, dated November 2, 2020, by and between NanoVibronix, Inc. and Thomas Mika (incorporated by reference
to Exhibit 10.6 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on November 5, 2020). |
| |
|
|
| 10.51 |
|
Form of Securities Purchase Agreement, dated December 2, 2020 (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 7, 2020). |
| |
|
|
| 10.52 |
|
Form of Registration Rights Agreement, dated December 2, 2020 (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 7, 2020). |
| |
|
|
| 10.53# |
|
Amended and Restated Distribution Agreement for “Private Labeled” Products dated December 10, 2020 by and between NanoVibronix, Inc. and Ultra Pain Products Inc (incorporated by reference to Exhibit 10.58 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 15, 2021). |
| |
|
|
| 10.54+ |
|
Second Amendment to the NanoVibronix, Inc. 2014 Long-Term Incentive Plan. (incorporated by reference to Annex A to the Company’s definitive proxy statement on Schedule 14A filed with the Securities and Exchange Commission on April 30, 2019). |
| |
|
|
| 10.55+ |
|
Third Amendment to the NanoVibronix, Inc. 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2021). |
| |
|
|
| 10.56+ |
|
Fourth Amendment to the NanoVibronix, Inc. 2014 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 15, 2022). |
| |
|
|
| 10.57
|
|
Form of Securities Purchase Agreement, dated November 29, 2022 (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 1, 2022). |
| |
|
|
| 10.58 |
|
Form of Securities Purchase Agreement, dated August 30, 2023 (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on September 1, 2023). |
| |
|
|
| 10.59 |
|
Form of Registration Rights Agreement, dated August 30, 2023 (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on September 1, 2023). |
| |
|
|
| 10.60+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Aurora Cassirer (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023).
|
| |
|
|
| 10.61+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Brian Murphy (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023). |
| |
|
|
| 10.62+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Christopher Fashek (incorporated by reference to Exhibit 10.3 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023).
|
| 10.63+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Harold Jacob (incorporated by reference to Exhibit 10.4 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023). |
| |
|
|
| 10.64+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Maria Schroeder (incorporated by reference to Exhibit 10.5 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023).
|
| |
|
|
| 10.65+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Martin Goldstein (incorporated by reference to Exhibit 10.6 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023).
|
| |
|
|
| 10.66+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Michael Ferguson (incorporated by reference to Exhibit 10.7 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023).
|
| |
|
|
| 10.67+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Stephen Brown (incorporated by reference to Exhibit 10.8 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023). |
| |
|
|
| 10.68+ |
|
Option Cancellation and Release Agreement, dated November 29, 2023, by and between NanoVibronix, Inc. and Thomas Mika (incorporated by reference to Exhibit 10.9 to the Current Report on Form 8-K filed with the Securities Exchange Commission on December 4, 2023). |
| |
|
|
| 10.69 |
|
Second Amendment to the Amended and Restated Distribution Agreement for “Private-Labled” Products dated December 10, 2020 by and between NanoVibronix, Inc. and Ultra Pain Products Inc. (incorporated by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 13, 2023). |
| |
|
|
| 10.70# |
|
Standalone Services Agreement, dated March 22, 2024, by and between NanoVibronix, Inc. and Veranex, Inc. (incorporated by reference to Exhibit 10.75 to the Annual Report on Form 10-K filed on April 8, 2024). |
| |
|
|
| 10.71 |
|
Research Agreement, dated October 1, 2023, by and between NanoVibronix Inc. and the Regents of the University of Michigan (incorporated by reference to Exhibit 10.76 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 8, 2024). |
| |
|
|
| 10.72+ |
|
Employment Agreement, dated as of September 20, 2024, by and between Brian Murphy and NanoVibronix, Inc. (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on September 25, 2024). |
| |
|
|
| 10.73+ |
|
NanoVibronix, Inc. Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 20, 2024). |
| |
|
|
| 10.74 |
|
Form of Exchange Agreement, effective as of January 7, 2025 (incorporated by reference to the Current Report on Form 8-K filed with the Securities and Exchange Commission on January 7, 2025). |
| |
|
|
| 10.75 |
|
Form of Securities Purchase Agreement, dated as of February 13, 2025, by and between NanoVibronix, Inc. and the purchaser named therein (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 14, 2025). |
| 10.76 |
|
Form of Registration Rights Agreement, dated as of February 13, 2025, by and between NanoVibronix, Inc. and the purchaser named therein (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 14, 2025). |
| |
|
|
| 10.77 |
|
Amended and Restated Senior Convertible Debenture Due the Earlier of the Trigger Date and November 13, 2025 (incorporated by reference to Exhibit 10.78 to the Annual Report on Form 10-K filed on March 31, 2025). |
| |
|
|
| 10.78 |
|
Consolidated Secured Note issued by ENvue Medical Holdings LLC to Alpha Capital Anstalt on January 17, 2025 (incorporated by reference to Exhibit 10.80 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on February 14, 2025). |
| |
|
|
| 10.79 |
|
Amendment to Consolidated Secured Note (incorporated by reference to Exhibit 10.82 to the Registration Statement on Form S-1 filed with the Securities and Exchange Commission on February 14, 2025). |
| |
|
|
| 10.80 |
|
Guaranty, dated as of April 11, 2025, by and between the Company and Alpha Capital Anstalt (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on April 11, 2025). |
| |
|
|
| 10.81 |
|
Consolidated Secured Note issued by ENvue Medical Holdings LLC to Alpha Capital Anstalt on January 28, 2025 (incorporated by reference to Exhibit 10.81 to the Company’s Registration Statement on Form S-1/A, as filed with the SEC on May 12, 2025). |
| |
|
|
| 10.82 |
|
Form of Amendment Agreement, dated as of May 12, 2025, by and among the Company and the holders party thereto (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the SEC on May 16, 2025). |
| |
|
|
| 10.83 |
|
Form of Securities Purchase Agreement, dated as of July 18, 2025, by and between the Company and the purchaser named therein (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on July 22, 2025). |
| |
|
|
| 10.84+ |
|
Employment Agreement, dated as of February 12, 2025, by and between Rita Silberberg and ENvue Medical Israel Ltd. (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on July 30, 2025). |
| |
|
|
| 10.85+ |
|
Amended and Restated Employment Agreement, dated as of August 11, 2025, by and between NanoVibronix, Inc. and Stephen Brown (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on August 14, 2025). |
| |
|
|
| 10.86 |
|
Form of Securities Purchase Agreement, dated September 17, 2025, by and between the Company and the purchaser thereto (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on September 17, 2025). |
| |
|
|
| 10.87+ |
|
First Amendment to the NanoVibronix, Inc. 2024 Long-Term Incentive Plan (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 5, 2025). |
| |
|
|
| 10.88+ |
|
Amended and Restated Employment Agreement, dated as of December 17, 2025, by and between the Company and Doron Besser, M.D. (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 23, 2025). |
| |
|
|
| 10.89# |
|
Consulting Agreement, dated as of December 18, 2025, by and between the Company and RCM Financial Consulting, Inc. (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 23, 2025). |
| |
|
|
| 10.90+ |
|
Amended and Restated 2024 Long Term Incentive Plan of ENvue Medical, Inc., dated as of December 24, 2025 (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on December 30, 2025). |
| |
|
|
| 10.91 |
|
Form of Amendment Agreement, dated as of January 30, 2026, by and among the Company and the holders party thereto (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on January 30, 2026). |
| |
|
|
| 10.92+ |
|
First Amendment, dated February 2, 2026, to the Amended and Restated Employment Agreement dated as of December 17, 2025, by and between the Company and Doron Besser (incorporated by reference to Exhibit 10.1 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 6, 2026). |
| |
|
|
| 10.93+ |
|
Chairman Agreement, dated February 2, 2026, by and between the Company and David Johnson (incorporated by reference to Exhibit 10.2 to the Current Report on Form 8-K filed with the Securities and Exchange Commission on February 6, 2026). |
| |
|
|
| 21.1 |
|
List
of Subsidiaries (incorporated by reference to Exhibit 21.1 to the Company’s Annual
Report on Form 10-K filed with the Securities and Exchange Commission on March 31, 2025). |
| |
|
|
| 23.1* |
|
Consent of Kost Forer Gabbay & Kasierer, a member of Ernst & Young Global. |
| |
|
|
| 31.1* |
|
Certification of Chief Executive Officer Pursuant to Section 302 of Sarbanes-Oxley Act of 2002 |
| |
|
|
| 31.2* |
|
Certification of Chief Financial Officer Pursuant to Section 302 of Sarbanes-Oxley Act of 2002 |
| |
|
|
| 32.1** |
|
Certification of Chief Executive Officer Pursuant to Section 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| |
|
|
| 32.2** |
|
Certification of Chief Financial Officer Pursuant to Section 18 U.S.C. Section 1350, as Adopted Pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| |
|
|
| 97.1 |
|
Compensation Recovery Policy, adopted by the Board of Directors on November 6, 2023 (incorporated by reference to Exhibit 97.1 to the Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 8, 2024). |
| 101.INS* |
|
Inline
XBRL Instance Document. |
| |
|
|
| 101.SCH* |
|
Inline
XBRL Taxonomy Extension Schema Document. |
| |
|
|
| 101.CAL* |
|
Inline
XBRL Taxonomy Extension Calculation Linkbase Document. |
| |
|
|
| 101.DEF* |
|
Inline
XBRL Taxonomy Extension Definition Linkbase Document. |
| |
|
|
| 101.LAB* |
|
Inline
XBRL Taxonomy Extension Labels Linkbase Document. |
| |
|
|
| 101.PRE* |
|
Inline
XBRL Taxonomy Extension Presentation Linkbase Document. |
| |
|
|
| 104 |
|
Cover
Page Interactive Data File (embedded within the Inline XBRL document). |
| * |
Filed
herewith. |
| ** |
Furnished
herewith. |
| + |
Management
contract or compensatory plan or arrangement. |
| # |
Portions
of this exhibit have been omitted pursuant to Item 601(b)(10)(iv) of Regulation S-K under the Securities Act of 1933, as amended,
because they are both (i) not material and (ii) the type that the registrant treats as private or confidential. A copy of the omitted
portions will be furnished to the Securities and Exchange Commission upon its request. |
SIGNATURES
Pursuant
to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed
on its behalf by the undersigned, thereunto duly authorized.
| |
ENVUE MEDICAL,
INC. |
| |
|
| |
By: |
/s/
Doron Besser |
| |
|
Doron
Besser |
| |
|
Chief
Executive Officer (Principal Executive Officer) |
| |
|
|
| |
Date: |
April 15, 2026 |
| |
|
|
| |
By: |
/s/
Nicole Fernandez-McGovern |
| |
|
Nicole
Fernandez-McGovern |
| |
|
Chief
Financial Officer (Principal Financial Officer) |
| |
|
|
| |
Date: |
April 15, 2026 |
Pursuant
to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the
Registrant and in the capacities and on the dates indicated.
| Signature |
|
Title |
|
Date |
| |
|
|
|
|
| /s/
DORON BESSER, M.D. |
|
Chief
Executive Officer and Director |
|
April 15, 2026 |
| Doron
Besser, M.D. |
|
(principal
executive officer) |
|
|
| |
|
|
|
|
| /s/
NICOLE FERNANDEZ-MCGOVERN |
|
Chief
Financial Officer |
|
April 15, 2026 |
| Nicole
Fernandez-McGovern |
|
(principal
financial officer) |
|
|
| |
|
|
|
|
| /s/
RITA SILBERBERG |
|
Chief
Accounting Officer |
|
April 15, 2026 |
| Rita
Silberberg |
|
(principal
accounting officer) |
|
|
| |
|
|
|
|
| /s/
DAVID JOHNSON |
|
Director |
|
April 15, 2026 |
| David
Johnson |
|
|
|
|
| |
|
|
|
|
| /s/
ZEEV ROTSTEIN |
|
Director |
|
April 15, 2026 |
| Zeev
Rotstein, M.D. |
|
|
|
|
| |
|
|
|
|
| /s/
NINO PIONATI |
|
Director |
|
April 15, 2026 |
| Nino
Pionati |
|
|
|
|
| |
|
|
|
|
| /s/
ALISON BURGETT |
|
Director |
|
April 15, 2026 |
| Alison Burgett |
|
|
|
|