
J.P. Morgan Presentation January 12, 2026

Forward Looking Statements and Disclaimer The presentation contains
forward-looking statements. Statements made or presented may include statements that are not historical facts and are considered forward-looking within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the
Securities Exchange Act of 1934, as amended. Words such as “believe,” “anticipate,” “plan,” “expect,” “intend,” “will,” “may,” “goal,” “potential,” “should,” “could,” “aim,” “estimate,” “predict,” “continue” and similar expressions or the
negative of these terms or other comparable terminology are intended to identify forward-looking statements, though not all forward-looking statements necessarily contain these identifying words. We intend these forward-looking statements to be
covered by the safe harbor provisions for forward-looking statements contained in Section 27A of the Securities Act and Section 21E of the Exchange Act. These forward-looking statements, including express and implied statements relating to the
preliminary and unaudited estimate of cash resources as of December 31, 2025 and the preliminary and unaudited estimate of net product revenue for Attruby for the quarter ended December 31, 2025; the commercial success of Attruby; the clinical
timeline and clinical and therapeutic potential for the new antibody depleter program for ATTR-CM; the timing and expectations regarding the status and progress of our various clinical trials, including data readouts for these trials; the
safety, efficacy and mechanisms and the clinical, therapeutic and market potential of our clinical development programs and our pipeline, including infigratinib, BBP-418, encaleret and BBP-812; expected timing for submitting New Drug
Applications with the U.S. Food and Drug Administration (“FDA”) and similar submissions with foreign regulatory authorities, receiving U.S. approval and commencing commercial launch for BBP-418 and encaleret; our anticipated interactions with
and feedback from the FDA and similar foreign regulatory authorities; the efficiency of our engine to rapidly and efficiently deliver medicines; our value creation potential for patients; the timing and progress of advancing GondolaBio’s
pipeline, including the clinical potential of GondolaBio’s PORT-77 in EPP; our financial position, including our expectations regarding potential market opportunities, and reaching certain clinical and regulatory milestones; the potency and
safety of our product candidates, the potential benefits of our product candidates; and the potential for greater patient access to medications, reflect our current views about our plans, intentions, expectations and strategies, which are based
on the information currently available to us and on assumptions we have made. Such statements reflect the current views of the Company with respect to future events and are subject to known and unknown risks, including business, regulatory,
economic and competitive risks, uncertainties, contingencies and assumptions about the Company, including, without limitation, risks inherent in developing and commercializing therapeutic products, and those risks and uncertainties described
under the heading “Risk Factors” in the Company’s most recent Annual Report on Form 10-K filed with the U.S. Securities and Exchange Commission (“SEC”) and in subsequent filings made by the Company with the SEC, which are available on the SEC’s
website at www.sec.gov. In light of these risks and uncertainties, many of which are beyond the Company’s control, the events or circumstances referred to in the forward-looking statements, express or implied, may not occur. The actual results
may vary from the anticipated results and the variations may be material. You are cautioned not to place undue reliance on these forward-looking statements, which speak to the Company’s current beliefs and expectations only as of the date of
the presentation. Except as required by law, the Company disclaims any intention or responsibility for updating or revising any forward-looking statements made or presented at the presentation in the event of new information, future
developments or otherwise. This presentation discusses product candidates that are investigational only and have not yet been approved for marketing by the FDA or any comparable foreign regulatory authority. No representation is made as to the
safety or effectiveness of the product candidates for the therapeutic use for which such product candidates are being studied. Certain information communicated at the presentation may relate to or is based on studies, publications, surveys and
other data obtained from third-party sources and the Company’s own internal estimates and research. While the Company believes these third-party sources to be reliable as of the date of the presentation, it has not independently verified, and
makes no representation as to the adequacy, fairness, accuracy or completeness of, any information obtained from third-party sources. In addition, certain information to be communicated at the presentation involves a number of assumptions and
limitations, and there can be no guarantee as to the accuracy or reliability of such assumptions. Finally, such research has not been verified by any independent source. Such information is provided as of the date of the presentation and is
subject to change without notice. The Company has not verified, and will not verify, any part of this presentation, and the Company makes no representation or warranty, express or implied, as to the accuracy or completeness of the information
to be communicated at the presentation or as to the existence, substance or materiality of any information omitted from the presentation at the presentation. The Company disclaims any and all liability for any loss or damage (whether
foreseeable or not) suffered or incurred by any person or entity as a result of anything contained or omitted from this document or the related presentation and such liability is expressly disclaimed. This Presentation is for informational
purposes only. This Presentation shall not constitute an offer to sell or the solicitation of an offer to buy any securities, nor shall there be any sale of any securities in any state or jurisdiction in which such offer, solicitation or sale
would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction. 2
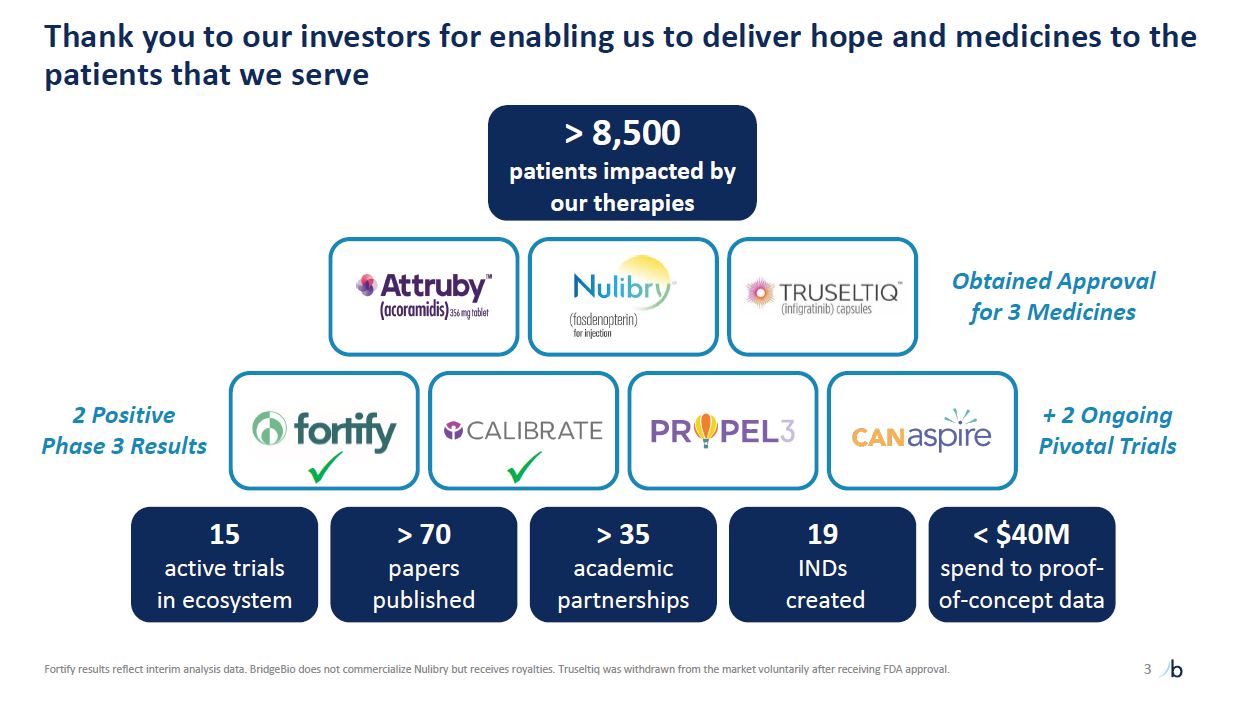
Thank you to our investors for enabling us to deliver hope and medicines to the
patients that we serve 3 Fortify results reflect interim analysis data. BridgeBio does not commercialize Nulibry but receives royalties. Truseltiq was withdrawn from the market voluntarily after receiving FDA approval. Obtained Approval for
3 Medicines + 2 Ongoing Pivotal Trials > 8,500 patients impacted by our therapies > 70 papers published > 35 academic partnerships 19 INDs created < $40M spend to proof-of-concept data 15 active trials in
ecosystem 2 Positive Phase 3 Results

Today we will review commercial/late-stage programs and early-stage
progress 4 Attruby revenue Late-stage data and regulatory progress Early-stage research and our potentially best-in-class EPP program

Continued Attruby commercial momentum 1Represents preliminary, unaudited results
for the fourth quarter ended December 31, 2025, based on management’s current expectations and subject to completion of year end audit procedures. See Forward Looking Statements and Disclaimer on slide 2 regarding risks and uncertainties that
could cause actual results to differ. $146M1 Q4 2025 Net Product Revenue NEW INFORMATION 5

Continued Attruby commercial momentum and patient impact 6 6,629 Unique U.S.
patient prescriptions for Attruby 1,632 Unique U.S. prescribing HCPs >25% Estimated share of NBRx for Attruby As of December 31st, 2025 NEW INFORMATION

We continue to study the impact of Attruby across clinical dimensions …approved
product with a label specifying near-complete stabilization of TTR First and ONLY… Reduction in cumulative cardiovascular outcomes within the first month of treatment in patients with ATTR-CM 1 month Reduction in composite of all-cause
mortality and recurrent cardiovascular-related hospitalization events at Month 30 42% Reduction in the cumulative frequency of cardiovascular-related hospitalization events at Month 30 50% 7
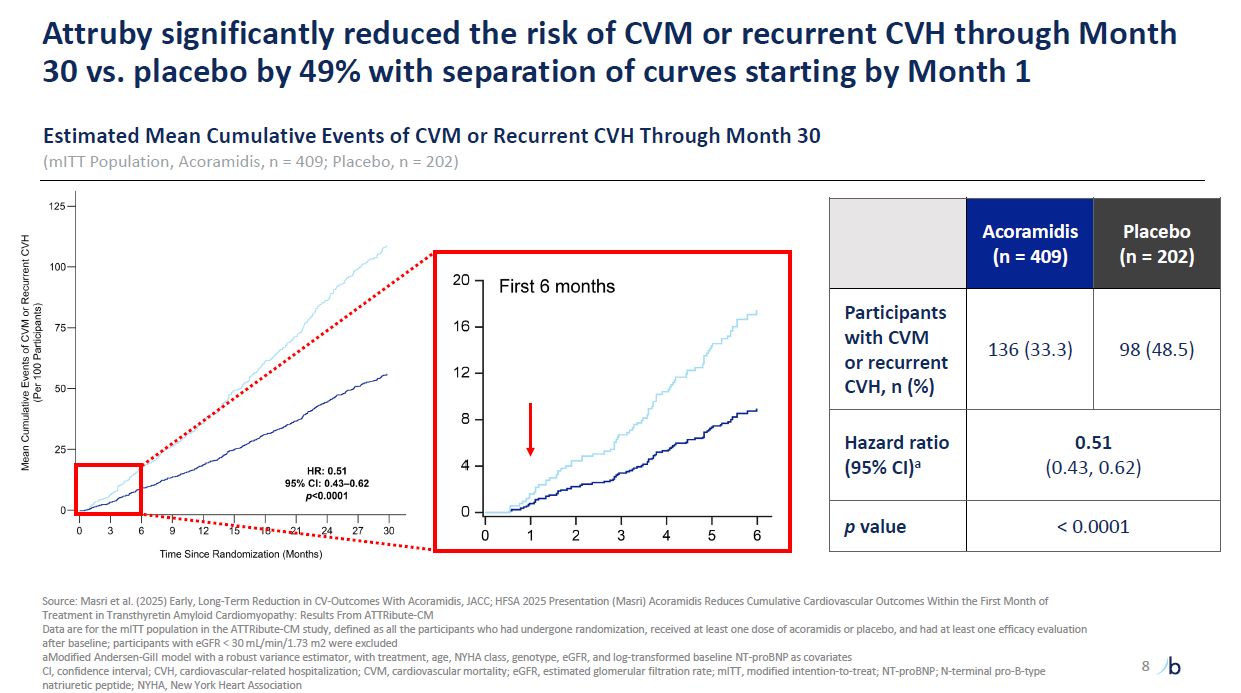
Attruby significantly reduced the risk of CVM or recurrent CVH through Month 30
vs. placebo by 49% with separation of curves starting by Month 1 Estimated Mean Cumulative Events of CVM or Recurrent CVH Through Month 30 (mITT Population, Acoramidis, n = 409; Placebo, n = 202) Acoramidis (n = 409) Placebo (n =
202) Participants with CVM or recurrent CVH, n (%) 136 (33.3) 98 (48.5) Hazard ratio (95% CI)a 0.51 (0.43, 0.62) p value < 0.0001 Source: Masri et al. (2025) Early, Long-Term Reduction in CV-Outcomes With Acoramidis, JACC; HFSA
2025 Presentation (Masri) Acoramidis Reduces Cumulative Cardiovascular Outcomes Within the First Month of Treatment in Transthyretin Amyloid Cardiomyopathy: Results From ATTRibute-CM Data are for the mITT population in the ATTRibute-CM study,
defined as all the participants who had undergone randomization, received at least one dose of acoramidis or placebo, and had at least one efficacy evaluation after baseline; participants with eGFR < 30 mL/min/1.73 m2 were
excluded aModified Andersen-Gill model with a robust variance estimator, with treatment, age, NYHA class, genotype, eGFR, and log-transformed baseline NT-proBNP as covariates CI, confidence interval; CVH, cardiovascular-related
hospitalization; CVM, cardiovascular mortality; eGFR, estimated glomerular filtration rate; mITT, modified intention-to-treat; NT-proBNP; N-terminal pro-B-type natriuretic peptide; NYHA, New York Heart Association 8

Attruby’s advantages stem from the fact that it is the most potent
stabilizer 9 Nelson et al., Amyloid 2020; 28(1), 24–29. Miller, M. et al. J Med Chem. 2018;61:7862-7876. Pinheiro, F. et al. Proc Natl Acad Sci U.S.A. 2026; 123(1) e2519908122. AG10 is the former internal development code for
Attruby/Beyonttra (acoramidis). Sees more target (superior free fraction) Binds more target (superior kd2) Glues the target together stronger (enthalpic binding mode) “Our thermodynamic analysis further supports the notion that binding
enthalpy (ΔH), not affinity (Kd or ΔG), better predicts the conformational stabilization imparted by kinetic stabilizers…. These results underscore the need to prioritize enthalpy-driven interactions during stabilizer design.” Acoramidis is
the only stabilizer with “near complete” stabilization in the label The multiplicity of advantages to superior stabilization continue to be better understood and elucidated in novel research “Given the variability in stoichiometry in the
experiments between tafamidis and AG10 and TTR, the data always tell the same story, that AG10 is better than tafamidis as would be expected from the determined binding constants.” – Prof. Jeffery Kelly (inventor of tafamidis) in email
correspondence with Dr. Isabella Graef, February 12, 2013. Bold added.
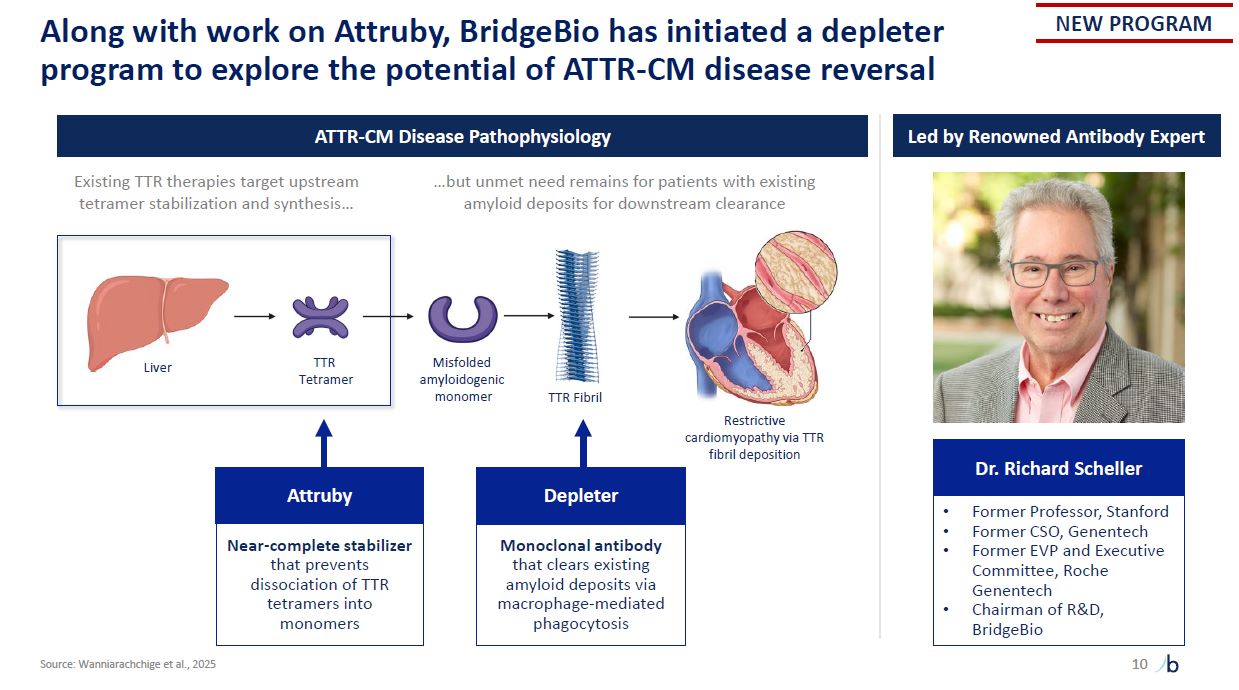
Liver TTR Tetramer Misfolded amyloidogenic monomer TTR Fibril Restrictive
cardiomyopathy via TTR fibril deposition Along with work on Attruby, BridgeBio has initiated a depleter program to explore the potential of ATTR-CM disease reversal 10 Source: Wanniarachchige et al., 2025 Attr uby Near-complete
stabilizer that prevents dissociation of TTR tetramers into monomers Depl eter Monoclonal antibody that clears existing amyloid deposits via macrophage-mediated phagocytosis Dr. Richard Scheller Former Professor, Stanford Former CSO,
Genentech Former EVP and Executive Committee, Roche Genentech Chairman of R&D, BridgeBio ATTR-CM Disease Pathophysiology Led by Renowned Antibody Expert NEW PROGRAM Existing TTR therapies target upstream …but unmet need remains for
patients with existing tetramer stabilization and synthesis… amyloid deposits for downstream clearance
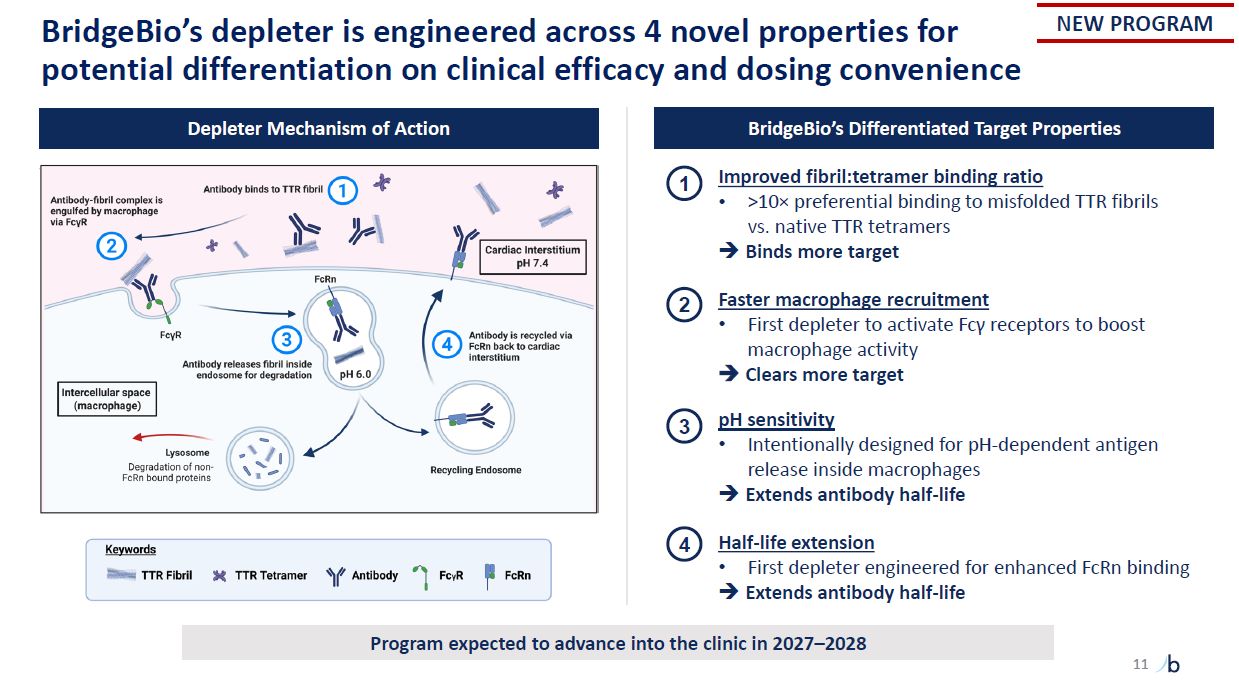
11 BridgeBio’s depleter is engineered across 4 novel properties for potential
differentiation on clinical efficacy and dosing convenience 1 2 3 4 Depleter Mechanism of Action BridgeBio’s Differentiated Target Properties Improved fibril:tetramer binding ratio >10× preferential binding to misfolded TTR fibrils
vs. native TTR tetramers Binds more target Faster macrophage recruitment First depleter to activate Fcγ receptors to boost macrophage activity Clears more target pH sensitivity Intentionally designed for pH-dependent antigen release
inside macrophages Extends antibody half-life Half-life extension First depleter engineered for enhanced FcRn binding Extends antibody half-life NEW PROGRAM Program expected to advance into the clinic in 2027–2028
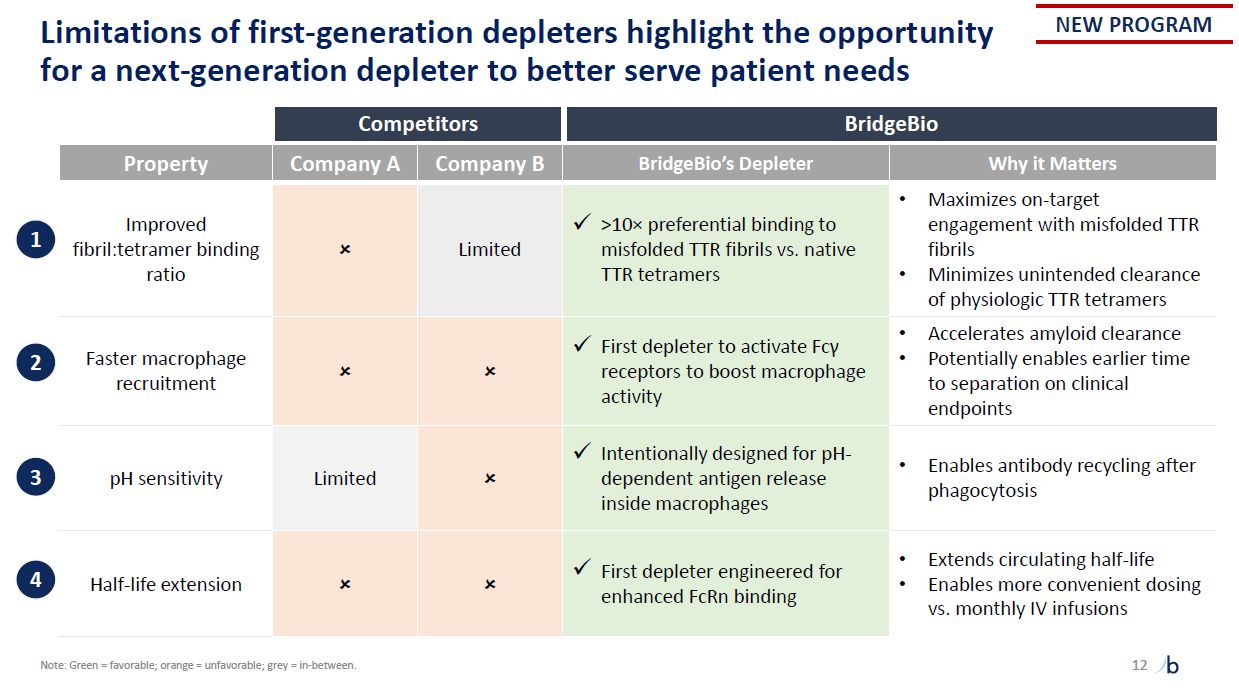
12 Limitations of first-generation depleters highlight the opportunity for a
next-generation depleter to better serve patient needs Competitors BridgeBio Property Company A Company B BridgeBio’s Depleter Why it Matters Improved fibril:tetramer binding ratio Limited >10× preferential binding to misfolded
TTR fibrils vs. native TTR tetramers Maximizes on-target engagement with misfolded TTR fibrils Minimizes unintended clearance of physiologic TTR tetramers Faster macrophage recruitment First depleter to activate Fcγ receptors to boost
macrophage activity Accelerates amyloid clearance Potentially enables earlier time to separation on clinical endpoints pH sensitivity Limited Intentionally designed for pH- dependent antigen release inside macrophages Enables antibody
recycling after phagocytosis Half-life extension First depleter engineered for enhanced FcRn binding Extends circulating half-life Enables more convenient dosing vs. monthly IV infusions 1 2 3 4 NEW PROGRAM Note: Green =
favorable; orange = unfavorable; grey = in-between.
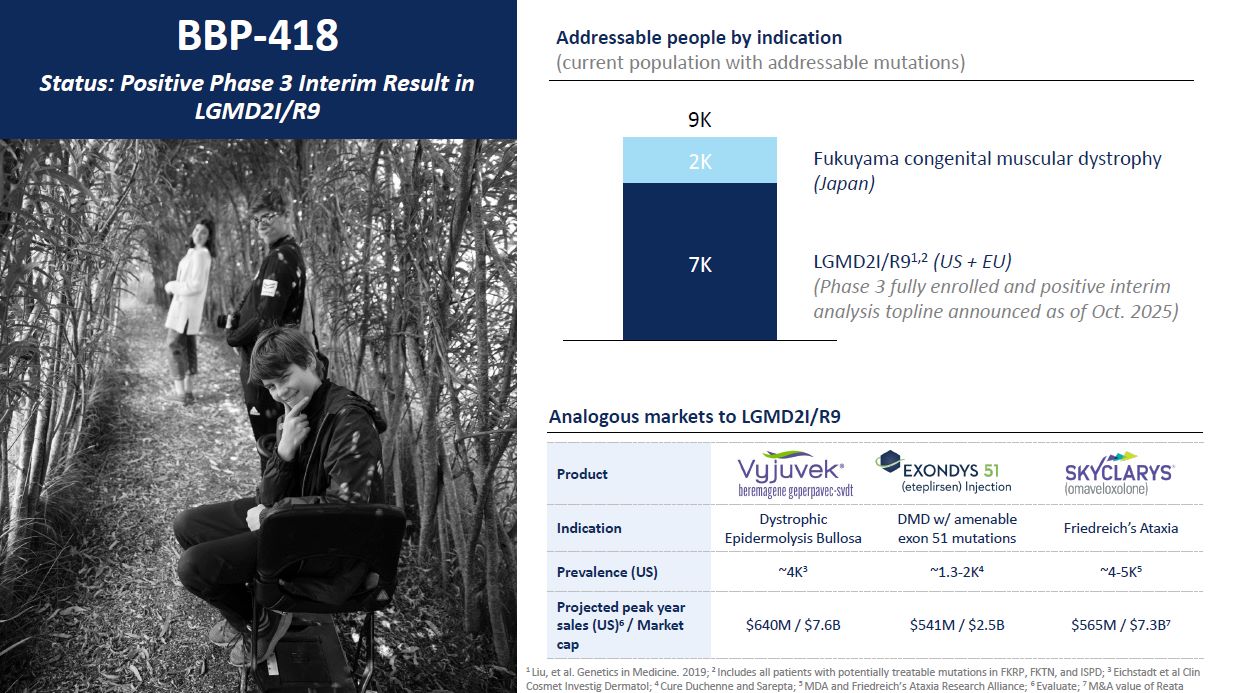
2K 7K 9K LGMD2I/R91,2 (US + EU) (Phase 3 fully enrolled and positive
interim analysis topline announced as of Oct. 2025) Fukuyama congenital muscular dystrophy (Japan) BBP-418 Status: Positive Phase 3 Interim Result in LGMD2I/R9 Addressable people by indication (current population with addressable
mutations) 1 Liu, et al. Genetics in Medicine. 2019; 2 Includes all patients with potentially treatable mutations in FKRP, FKTN, and ISPD; 3 Eichstadt et al Clin Cosmet Investig Dermatol; 4 Cure Duchenne and Sarepta; 5 MDA and Friedreich’s
Ataxia Research Alliance; 6 Evaluate; 7 M&A value of Reata Product Indication Dystrophic Epidermolysis Bullosa DMD w/ amenable exon 51 mutations Friedreich’s Ataxia Prevalence (US) ~4K3 ~1.3-2K4 ~4-5K5 Projected peak year sales
(US)6 / Market cap $640M / $7.6B $541M / $2.5B $565M / $7.3B7 Analogous markets to LGMD2I/R9
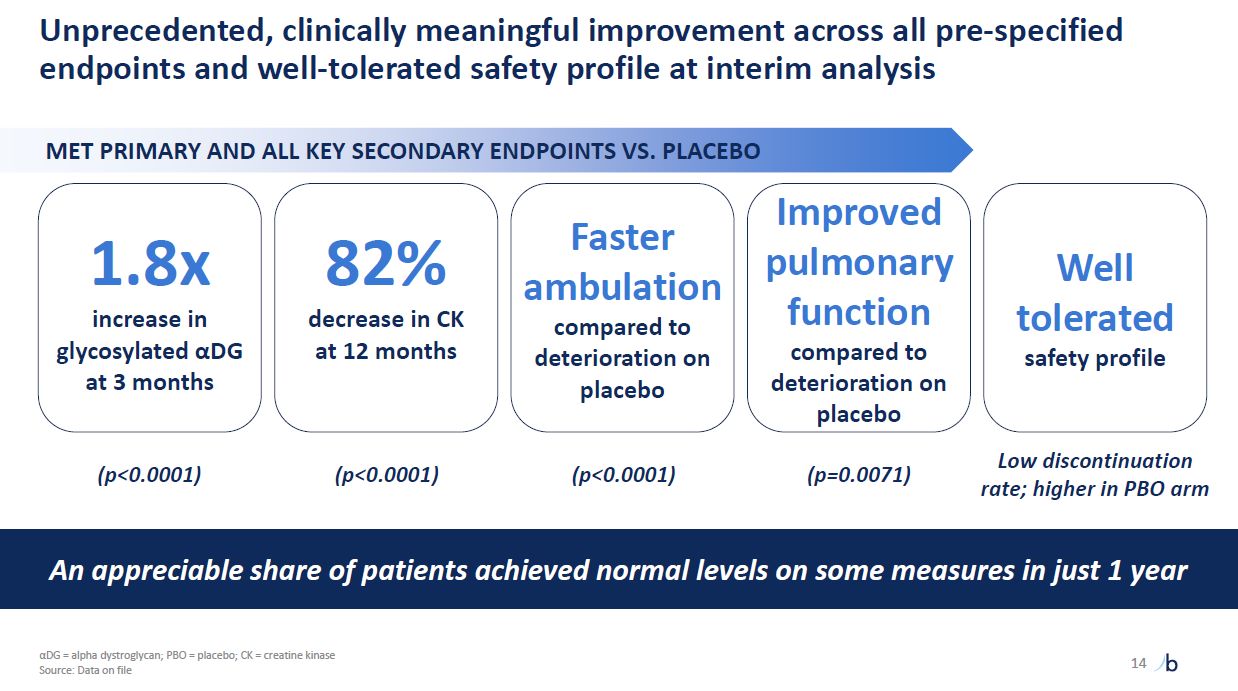
Unprecedented, clinically meaningful improvement across all pre-specified
endpoints and well-tolerated safety profile at interim analysis MET PRIMARY AND ALL KEY SECONDARY ENDPOINTS VS. PLACEBO 14 1.8x increase in glycosylated αDG at 3 months 82% decrease in CK at 12 months Faster ambulation compared to
deterioration on placebo Improved pulmonary function compared to deterioration on placebo (p<0.0001) An appreciable share of patients achieved normal levels on some measures in just 1 year Well tolerated safety
profile (p<0.0001) (p<0.0001) (p=0.0071) Low discontinuation rate; higher in PBO arm αDG = alpha dystroglycan; PBO = placebo; CK = creatine kinase Source: Data on file
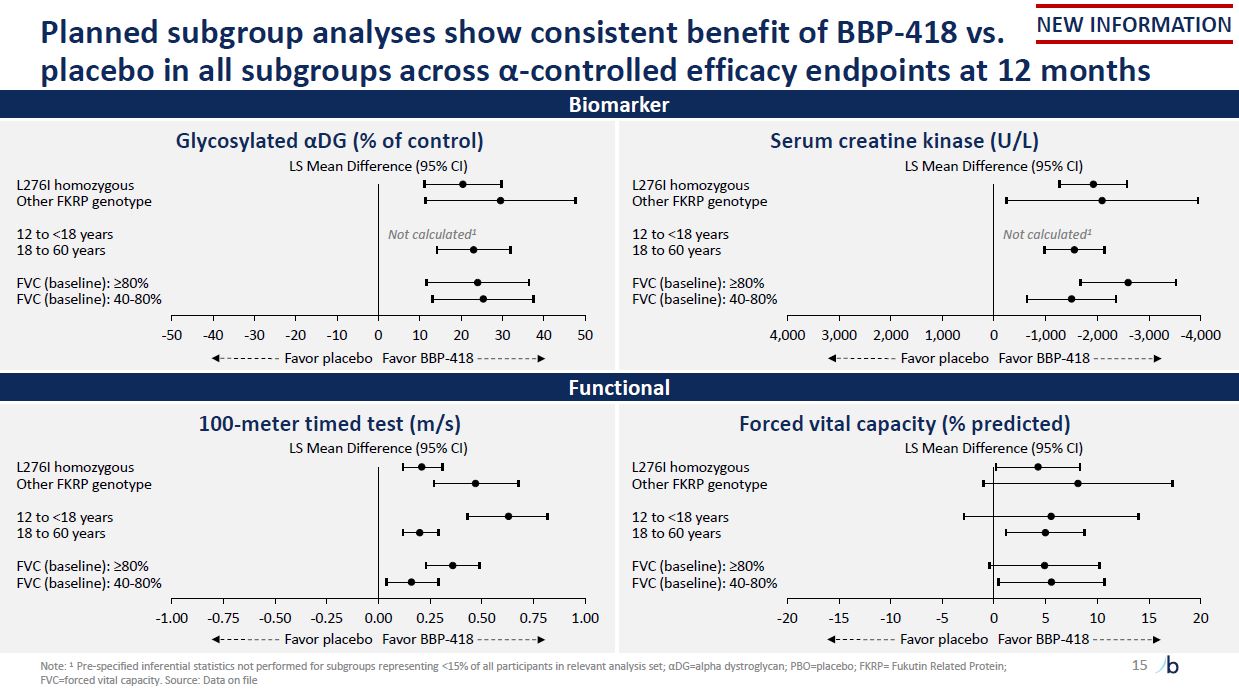
-20 -15 10 15 20 L276I homozygous Other FKRP genotype 12 to <18 years 18
to 60 years FVC (baseline): ≥80% FVC (baseline): 40-80% -1.00 -0.75 -0.50 -0.25 0.00 0.25 0.50 0.75 1.00 L276I homozygous Other FKRP genotype 12 to <18 years 18 to 60 years FVC (baseline): ≥80% FVC (baseline): 40-80% Planned
subgroup analyses show consistent benefit of BBP-418 vs. placebo in all subgroups across α-controlled efficacy endpoints at 12 months 15 Biomarker Forced vital capacity (% predicted) LS Mean Difference (95% CI) 100-meter timed test
(m/s) LS Mean Difference (95% CI) Functional Note: 1 Pre-specified inferential statistics not performed for subgroups representing <15% of all participants in relevant analysis set; αDG=alpha dystroglycan; PBO=placebo; FKRP= Fukutin
Related Protein; FVC=forced vital capacity. Source: Data on file Glycosylated αDG (% of control) LS Mean Difference (95% CI) L276I homozygous Other FKRP genotype 12 to <18 years Not calculated1 18 to 60 years FVC (baseline): ≥80% FVC
(baseline): 40-80% -50 -40 -30 -20 -10 0 10 20 30 40 50 Favor placebo Favor BBP-418 Serum creatine kinase (U/L) LS Mean Difference (95% CI) L276I homozygous Other FKRP genotype 12 to <18 years Not calculated1 18 to 60 years FVC
(baseline): ≥80% FVC (baseline): 40-80% 4,000 3,000 2,000 1,000 0 -1,000 -2,000 -3,000 -4,000 Favor placebo Favor BBP-418 -10 -5 0 5 Favor placebo Favor BBP-418 Favor placebo Favor BBP-418 NEW INFORMATION
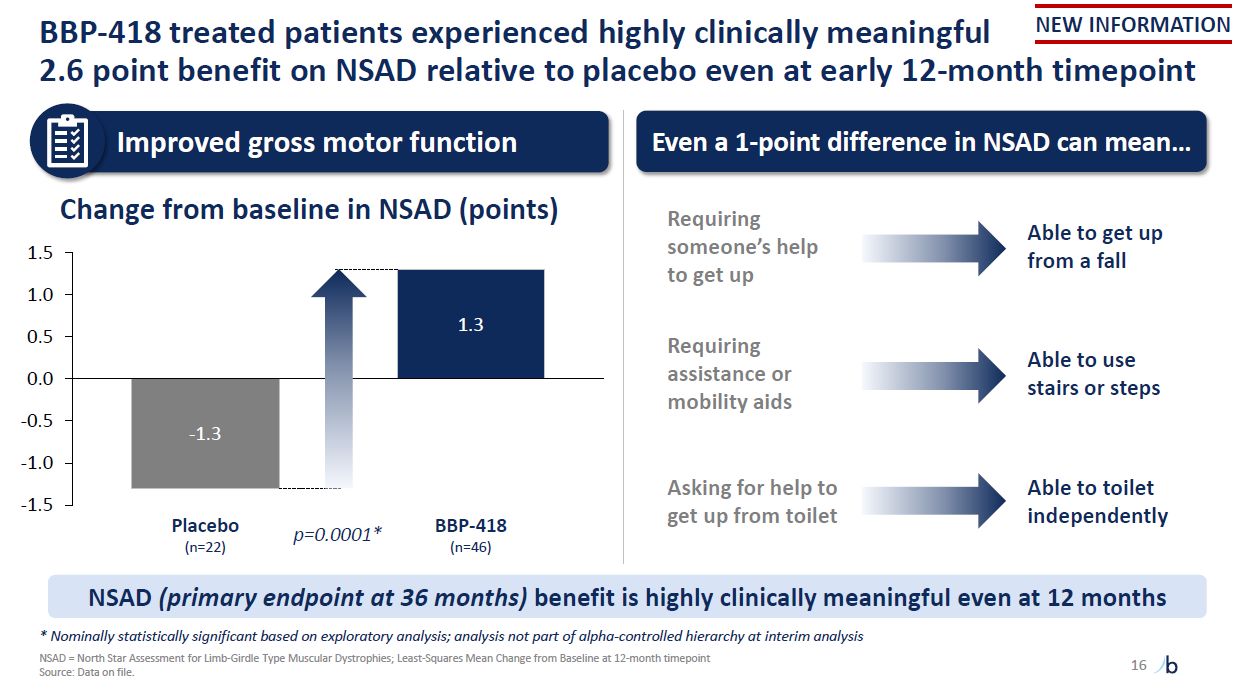
BBP-418 treated patients experienced highly clinically meaningful 2.6 point
benefit on NSAD relative to placebo even at early 12-month timepoint 16 -1.3 1.3 -1.5 -1.0 -0.5 0.0 0.5 1.0 1.5 Placebo (n=22) BBP-418 (n=46) p=0.0001* Change from baseline in NSAD (points) Source: Data on file. Improved gross
motor function Even a 1-point difference in NSAD can mean… Requiring someone’s help to get up Able to get up from a fall Requiring assistance or mobility aids Able to use stairs or steps Able to toilet independently Asking for help to
get up from toilet NSAD (primary endpoint at 36 months) benefit is highly clinically meaningful even at 12 months * Nominally statistically significant based on exploratory analysis; analysis not part of alpha-controlled hierarchy at interim
analysis NSAD = North Star Assessment for Limb-Girdle Type Muscular Dystrophies; Least-Squares Mean Change from Baseline at 12-month timepoint NEW INFORMATION

We completed a successful meeting with the FDA and they 17 We anticipate filing
an NDA with the FDA in 1H 2026 recommended orienting our NDA toward traditional and full approval All data from the Phase 3 FORTIFY study were presented to the agency, including key sensitivities FDA acknowledged the data “…demonstrate
consistent treatment effects on multiple efficacy endpoints” FDA recommends orienting NDA toward traditional approval NEW INFORMATION
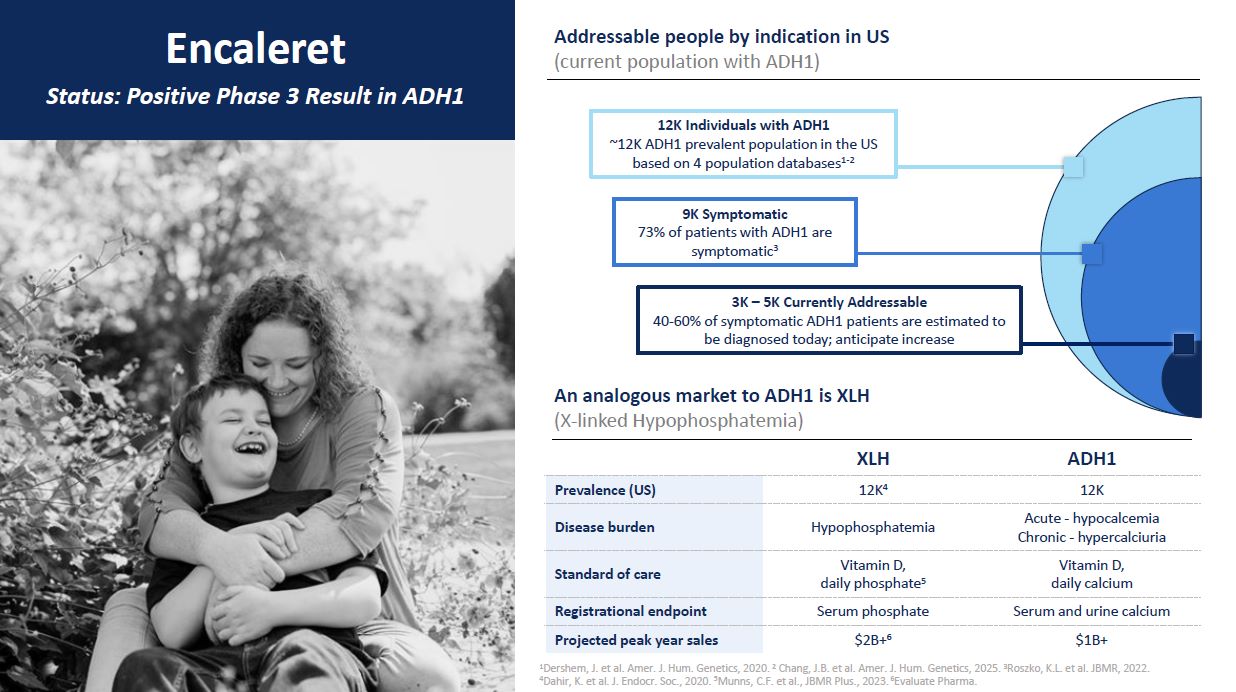
Encaleret Status: Positive Phase 3 Result in ADH1 Addressable people by
indication in US (current population with ADH1) An analogous market to ADH1 is XLH (X-linked Hypophosphatemia) XLH ADH1 Prevalence (US) 12K4 12K Disease burden Hypophosphatemia Acute - hypocalcemia Chronic - hypercalciuria Standard
of care Vitamin D, daily phosphate5 Vitamin D, daily calcium Registrational endpoint Serum phosphate Serum and urine calcium Projected peak year sales $2B+6 $1B+ 1Dershem, J. et al. Amer. J. Hum. Genetics, 2020. 2 Chang, J.B. et al.
Amer. J. Hum. Genetics, 2025. 3Roszko, K.L. et al. JBMR, 2022. 4Dahir, K. et al. J. Endocr. Soc., 2020. 5Munns, C.F. et al., JBMR Plus., 2023. 6Evaluate Pharma. 12K Individuals with ADH1 ~12K ADH1 prevalent population in the US based on 4
population databases1-2 9K Symptomatic 73% of patients with ADH1 are symptomatic3 3K – 5K Currently Addressable 40-60% of symptomatic ADH1 patients are estimated to be diagnosed today; anticipate increase

CALIBRATE achieved & exceeded all criteria set forth as an upside target,
with a 76% responder rate following 24 weeks of encaleret treatment Upside Target Clinical Profile Outcome Observed Statistically significant primary analysis result compared to conventional therapy Primary endpoint met (p <0.0001)
demonstrating superiority to conventional therapy At Week 24, ≥50% of study participants achieve target serum and urine Ca on encaleret 76% (34 out of 45) achieved target serum and urine Ca on encaleret vs. 4% on conventional
therapy Majority of participants randomized to encaleret able to remain independent from conventional therapy1 Among encaleret responders at Week 24, none required conventional therapy during Period 31 At Week 24, mean iPTH within normal
range on encaleret >90% of participants administered encaleret achieved iPTH above the lower limit of the reference range Comparable safety and tolerability profile to conventional therapy Encaleret was well-tolerated; no discontinuations
related to study drug 19 1Requirement for conventional therapy defined as oral calcium >600 mg/day and/or active vitamin D during Period 3. Ca = Calcium; iPTH = Intact Parathyroid Hormone. Encaleret is an investigational
drug. Its safety and efficacy have not been fully evaluated by any regulatory authority.
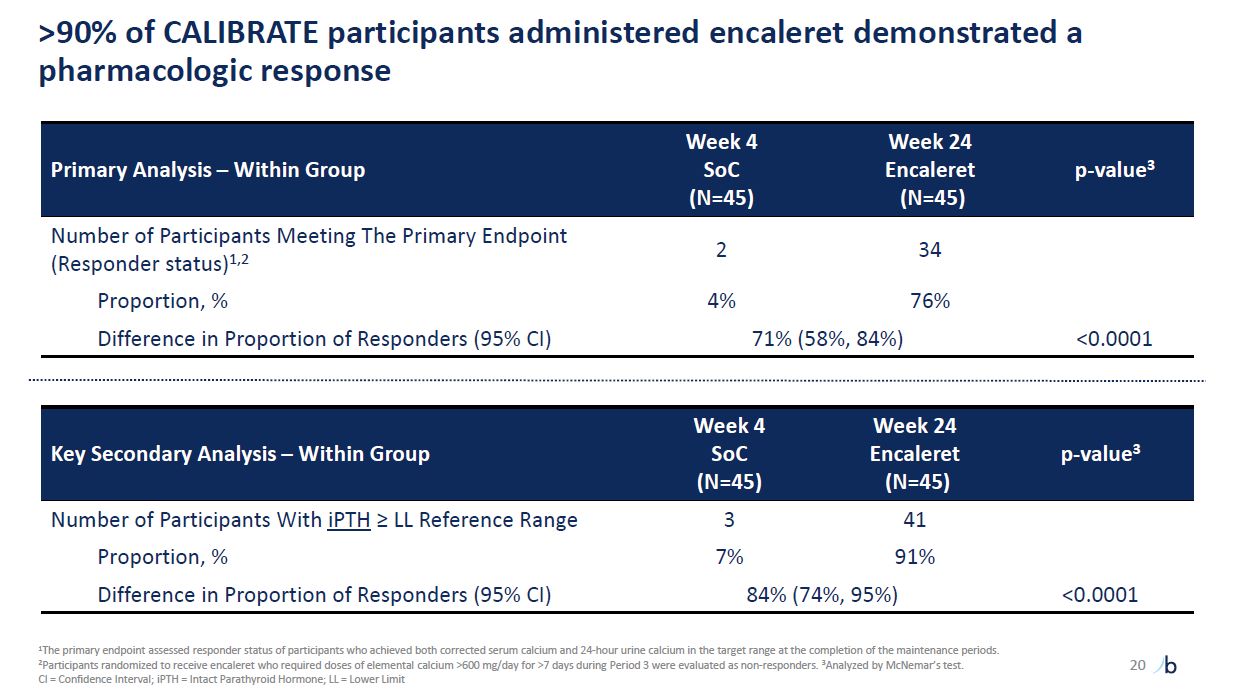
>90% of CALIBRATE participants administered encaleret demonstrated a
pharmacologic response Primary Analysis – Within Group Week 4 SoC (N=45) Week 24 Encaleret (N=45) p-value3 Number of Participants Meeting The Primary Endpoint (Responder status)1,2 2 34 Proportion, % 4% 76% Difference in Proportion
of Responders (95% CI) 71% (58%, 84%) <0.0001 Key Secondary Analysis – Within Group Week 4 SoC (N=45) Week 24 Encaleret (N=45) p-value3 Number of Participants With iPTH ≥ LL Reference Range 3 41 Proportion, % 7% 91% Difference
in Proportion of Responders (95% CI) 84% (74%, 95%) <0.0001 20 1The primary endpoint assessed responder status of participants who achieved both corrected serum calcium and 24-hour urine calcium in the target range at the completion of
the maintenance periods. 2Participants randomized to receive encaleret who required doses of elemental calcium >600 mg/day for >7 days during Period 3 were evaluated as non-responders. 3Analyzed by McNemar’s test. CI = Confidence
Interval; iPTH = Intact Parathyroid Hormone; LL = Lower Limit
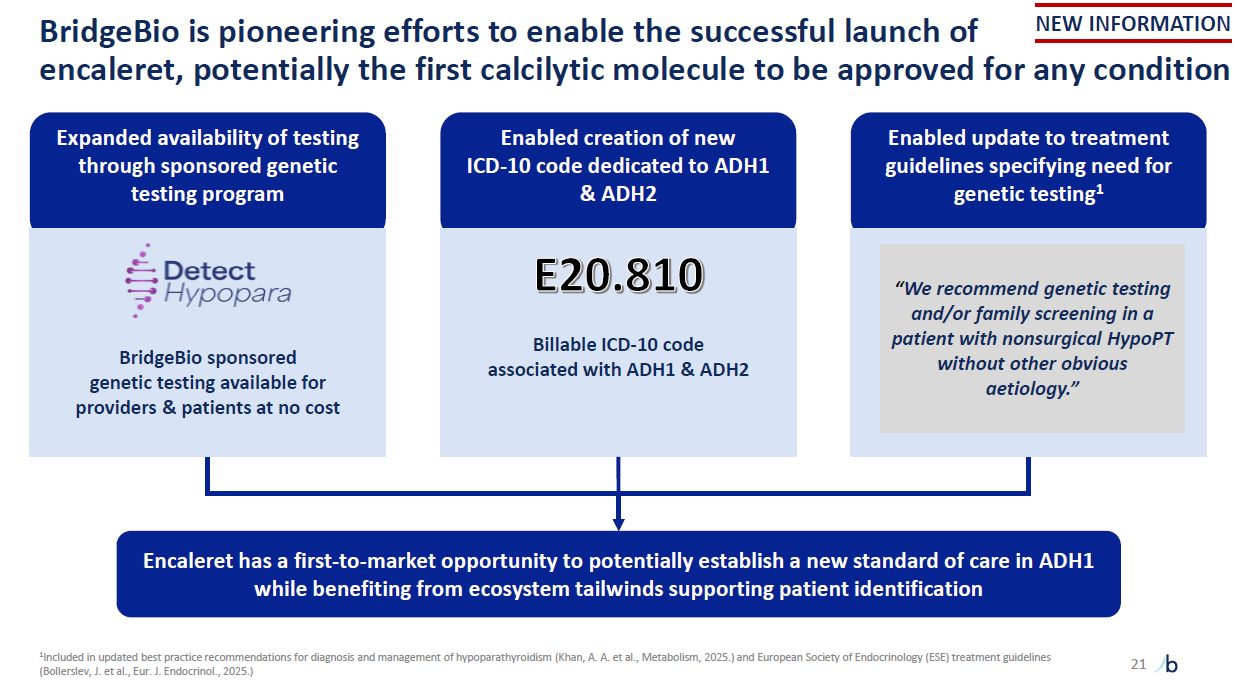
BridgeBio is pioneering efforts to enable the successful launch of encaleret,
potentially the first calcilytic molecule to be approved for any condition Enabled creation of new ICD-10 code dedicated to ADH1 & ADH2 Billable ICD-10 code associated with ADH1 & ADH2 Encaleret has a first-to-market opportunity to
potentially establish a new standard of care in ADH1 while benefiting from ecosystem tailwinds supporting patient identification Enabled update to treatment guidelines specifying need for genetic testing1 “We recommend genetic testing and/or
family screening in a patient with nonsurgical HypoPT without other obvious aetiology.” Expanded availability of testing through sponsored genetic testing program BridgeBio sponsored genetic testing available for providers & patients at
no cost 21 NEW INFORMATION 1Included in updated best practice recommendations for diagnosis and management of hypoparathyroidism (Khan, A. A. et al., Metabolism, 2025.) and European Society of Endocrinology (ESE) treatment guidelines
(Bollerslev, J. et al., Eur. J. Endocrinol., 2025.)
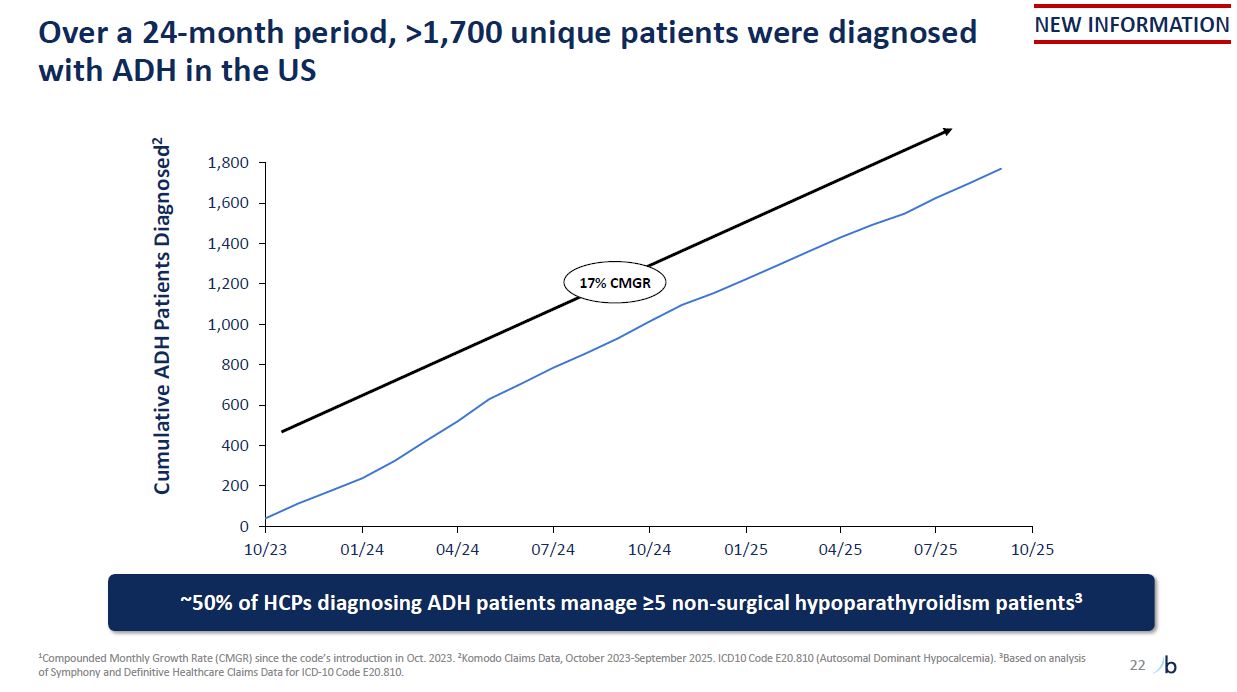
Over a 24-month period, >1,700 unique patients were diagnosed with ADH in the
US 1Compounded Monthly Growth Rate (CMGR) since the code’s introduction in Oct. 2023. 2Komodo Claims Data, October 2023-September 2025. ICD10 Code E20.810 (Autosomal Dominant Hypocalcemia). 3Based on analysis of Symphony and Definitive
Healthcare Claims Data for ICD-10 Code E20.810. Cumulative ADH Patients Diagnosed2 1,000 800 600 400 200 0 10/23 01/24 04/24 07/24 10/24 01/25 04/25 07/25 10/25 ~50% of HCPs diagnosing ADH patients manage ≥5 non-surgical
hypoparathyroidism patients3 1,200 1,800 1,600 1,400 17% CMGR NEW INFORMATION 22
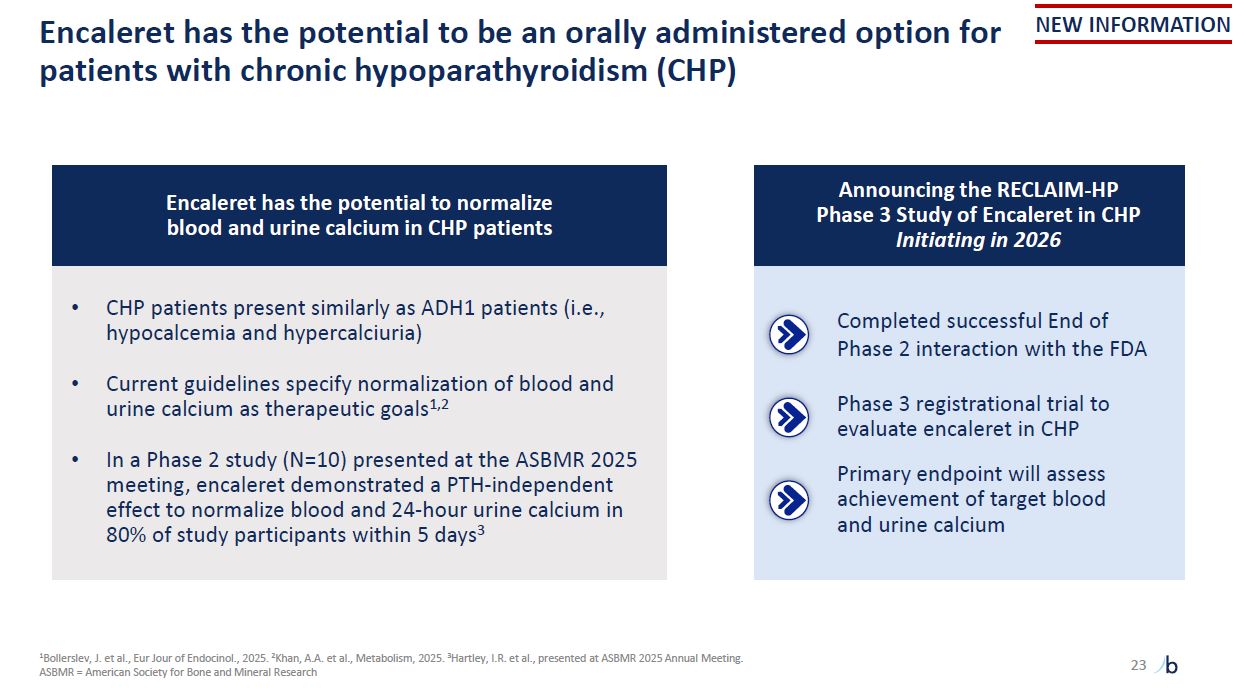
Encaleret has the potential to be an orally administered option for patients with
chronic hypoparathyroidism (CHP) 1Bollerslev, J. et al., Eur Jour of Endocinol., 2025. 2Khan, A.A. et al., Metabolism, 2025. 3Hartley, I.R. et al., presented at ASBMR 2025 Annual Meeting. ASBMR = American Society for Bone and Mineral
Research NEW INFORMATION 23 Encaleret has the potential to normalize blood and urine calcium in CHP patients CHP patients present similarly as ADH1 patients (i.e., hypocalcemia and hypercalciuria) Current guidelines specify normalization
of blood and urine calcium as therapeutic goals1,2 In a Phase 2 study (N=10) presented at the ASBMR 2025 meeting, encaleret demonstrated a PTH-independent effect to normalize blood and 24-hour urine calcium in 80% of study participants within
5 days3 Announcing the RECLAIM-HP Phase 3 Study of Encaleret in CHP Initiating in 2026 Completed successful End of Phase 2 interaction with the FDA Phase 3 registrational trial to evaluate encaleret in CHP Primary endpoint will assess
achievement of target blood and urine calcium
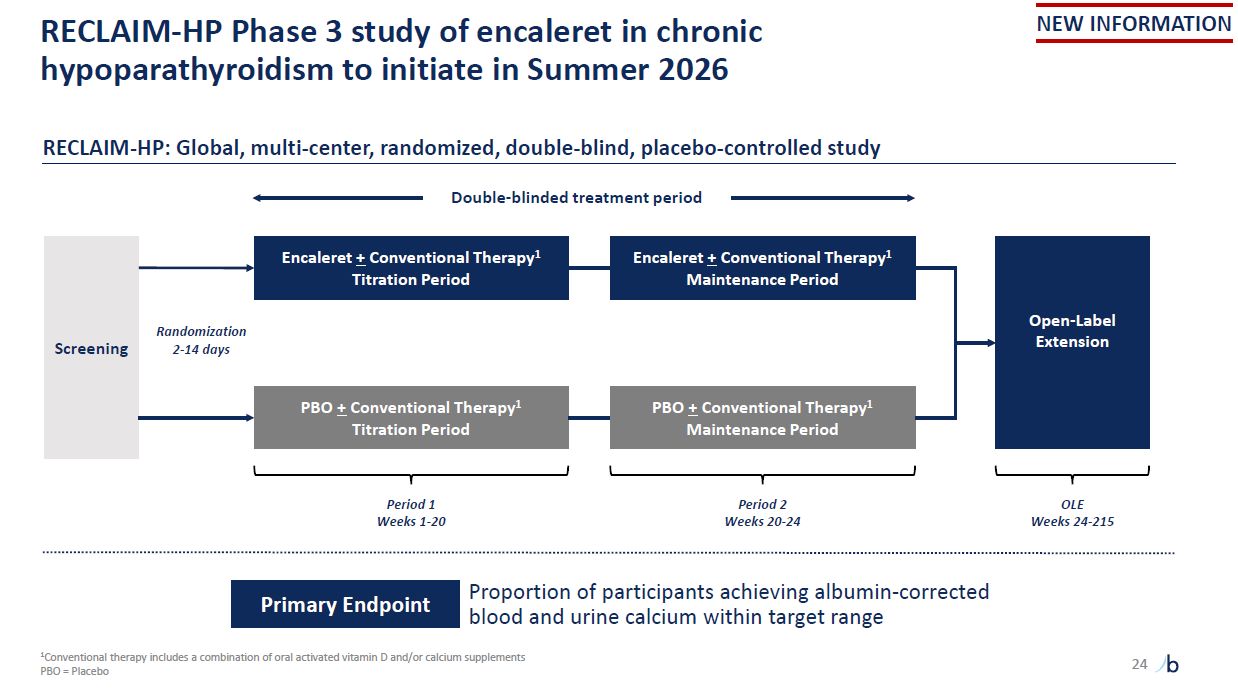
RECLAIM-HP Phase 3 study of encaleret in chronic hypoparathyroidism to initiate in
Summer 2026 1Conventional therapy includes a combination of oral activated vitamin D and/or calcium supplements PBO = Placebo NEW INFORMATION Screening Period 1 Weeks 1-20 Encaleret + Conventional Therapy1 Titration Period PBO +
Conventional Therapy1 Titration Period Encaleret + Conventional Therapy1 Maintenance Period PBO + Conventional Therapy1 Maintenance Period Open-Label Extension Period 2 Weeks 20-24 OLE Weeks 24-215 RECLAIM-HP: Global, multi-center,
randomized, double-blind, placebo-controlled study Double-blinded treatment period Proportion of participants achieving albumin-corrected blood and urine calcium within target range Primary Endpoint Randomization 2-14 days 24

Infigratinib Status: LPLV in Phase 3 in achondroplasia 1CDC birth estimates; EU
Eurostats birth estimates; Foreman, et al. Am J Med Genet. 2020.; Bober, et al. Gene Reviews. 2020.; Wenger, et al. Gene Reviews. 2020.; Al-Namman, et al. J Oral Biol Craniofac Res. 2019. 2Achondroplasia market includes all approved
drugs. Represents diagnosed and addressable ACH population with open growth plates 55,000 individuals with achondroplasia in US/EU $5B+ potential global market

Last participant last visit completed; Topline expected Q1 2026 Pivotal Phase 3
Study in children and adolescents (3-<18 years) with achondroplasia and open epiphyses Phase 2 Study In Infants & Toddlers from birth to less than age 3 with achondroplasia Full enrollment completed for Phase 2 portion; data expected
2H 2026 2/3 Phase 2 study followed by a Phase 3 study in children and adolescents with hypochondroplasia First participant enrolled NEW INFORMATION We have achieved LPLV on PROPEL 3 and expect topline in Q1, and we have made significant
operational progress on expansion opportunities 26

Designed to target achondroplasia at its genetic source: FGFR3
overactivation Addresses not just overactivation of the MAPK pathway (chondrocyte hypertrophy), but also STAT1 (chondrocyte proliferation) and all other downstream pathways Achieved profound efficacy in animal models, beyond just long bone
growth In mouse models of achondroplasia, treatment with infigratinib showed an increase in proximal and distal long bone length (femur +21%, humerus +12%, tibia +33%, ulna 22%, and radius +24%) and foramen magnum area (+17%)5 Demonstrated
the largest degree of efficacy (across multiple dimensions1) across any clinical trial for ACH2 Mean change from baseline in AHV: +2.51 cm/yr at M12 Mean absolute AHV: >6 cm/yr at M12 Mean change in height Z-score compared to ACH growth
charts: +0.36 SD at M12 Mean improvement in upper-to-lower body segment ratio (proportionality): Decrease of 0.12 (P=0.001) Received the only Breakthrough Designation from the FDA for ACH Met the regulatory requirement of showing preliminary
evidence of substantial improvement over SoC Designed to be taken as a daily oral, avoiding side effects associated with CNPs and repeated injections Avoids symptomatic hypotension1, injection site reactions1, and the psychosocial burden of
receiving/administering repeated injections3,4 27 Infigratinib: Defining characteristics of a potentially best-in-class program in the ACH landscape 1Savarirayan et al, NEJM, 2024; 2For monotherapy trials in ACH; 3Antal et al, Journal of
Pediatric Psychology, 2011; 4Jacobse et al, European Journal of Pediatrics, 2019; 5Komla-Ebri et al, J Clin Invest. 2016.

Infigratinib sprinkle capsules are being developed for oral
administration1,2 28 1Savarirayan R, et al. N Engl J Med. 2024;392(9):865–874. 2BridgeBio data on file. *Infigratinib is an investigational agent that is not approved for use by any regulatory authority. Size 2 capsules are shown in
photo. Infigratinib is being studied in children over 3 years of age with achondroplasia (0.25 kg/mg/day) as a sprinkle capsule Capsules can be swallowed whole or content (granules) sprinkled on soft food The dosage strength of each capsule
depends on how many granules are inside Each child’s dose is based on their weight Capsules (17 mm long*) Granules (2 mm long)
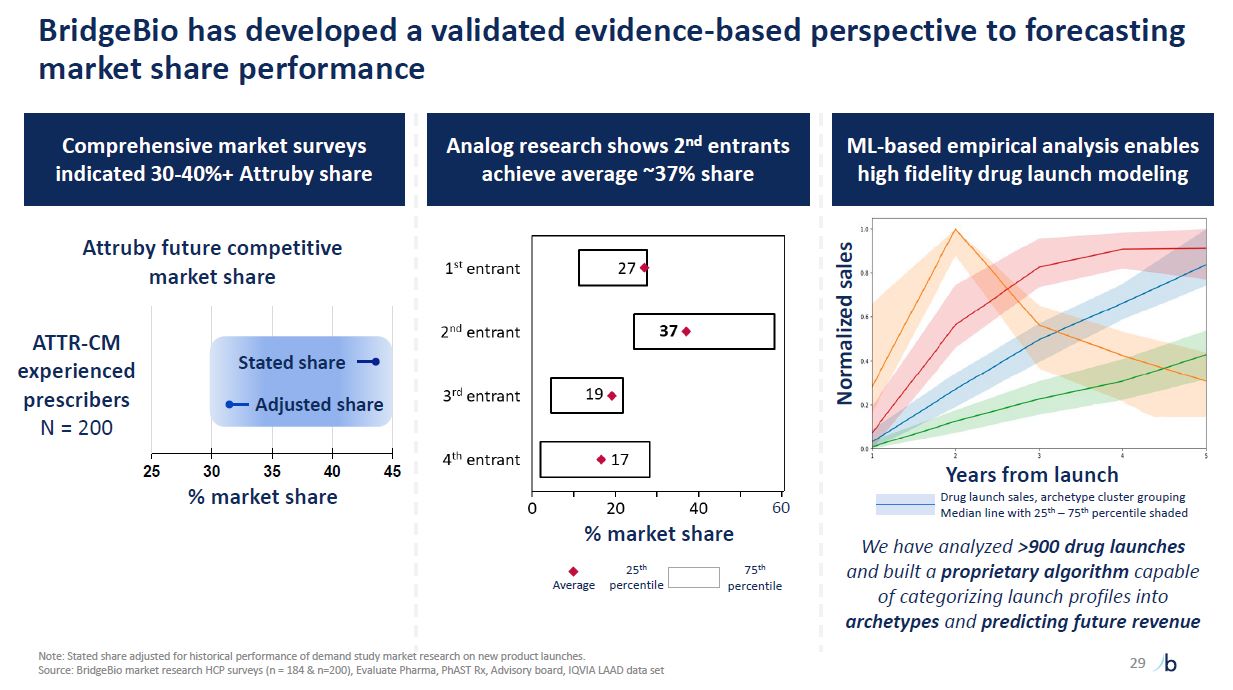
BridgeBio has developed a validated evidence-based perspective to forecasting
market share performance 29 Comprehensive market surveys indicated 30-40%+ Attruby share Attruby future competitive market share 25 30 35 40 % market share 45 ATTR-CM experienced prescribers N = 200 Stated share Adjusted
share 60 Analog research shows 2nd entrants achieve average ~37% share Average 25th percentile 75th percentile ML-based empirical analysis enables high fidelity drug launch modeling % market share Normalized sales Years from
launch Drug launch sales, archetype cluster grouping Median line with 25th – 75th percentile shaded We have analyzed >900 drug launches and built a proprietary algorithm capable of categorizing launch profiles into archetypes and
predicting future revenue Note: Stated share adjusted for historical performance of demand study market research on new product launches. Source: BridgeBio market research HCP surveys (n = 184 & n=200), Evaluate Pharma, PhAST Rx,
Advisory board, IQVIA LAAD data set
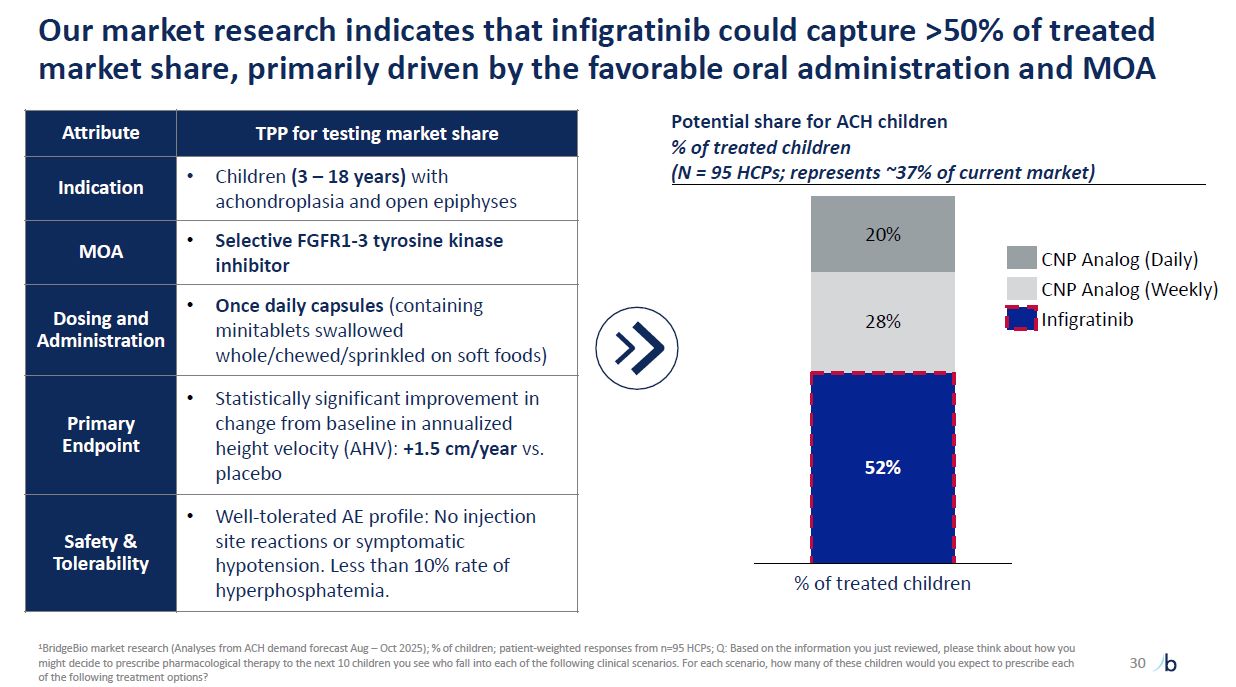
Our market research indicates that infigratinib could capture >50% of treated
market share, primarily driven by the favorable oral administration and MOA 30 Attribute TPP for testing market share Indication Children (3 – 18 years) with achondroplasia and open epiphyses MOA Selective FGFR1-3 tyrosine kinase
inhibitor Dosing and Administration Once daily capsules (containing minitablets swallowed whole/chewed/sprinkled on soft foods) Primary Endpoint Statistically significant improvement in change from baseline in annualized height velocity
(AHV): +1.5 cm/year vs. placebo Safety & Tolerability Well-tolerated AE profile: No injection site reactions or symptomatic hypotension. Less than 10% rate of hyperphosphatemia. Potential share for ACH children % of treated children (N
= 95 HCPs; represents ~37% of current market) 52% 28% 20% % of treated children CNP Analog (Daily) CNP Analog (Weekly) Infigratinib 1BridgeBio market research (Analyses from ACH demand forecast Aug – Oct 2025); % of children;
patient-weighted responses from n=95 HCPs; Q: Based on the information you just reviewed, please think about how you might decide to prescribe pharmacological therapy to the next 10 children you see who fall into each of the following clinical
scenarios. For each scenario, how many of these children would you expect to prescribe each of the following treatment options?

BBP-812 Status: Pivotal trial ongoing in Canavan disease Canavan disease (CD) is
an ultra-rare neurodegenerative disease with ~1,000 patients across the US and EU CD is usually fatal within the first two decades of life, and >25% of patients die by the age of 10 years1 Children with CD exhibit global and severe
cognitive, motor, and language impairment, missing or regressing on most developmental milestones Children with CD require around the clock care – they cannot hold their heads up, sit, crawl, walk, are generally unable to speak, and suffer
from seizures and spasticity There are no therapies available for Canavan disease Canavan disease is a fatal, neurodegenerative and ultra-rare pediatric disease with no approved therapies
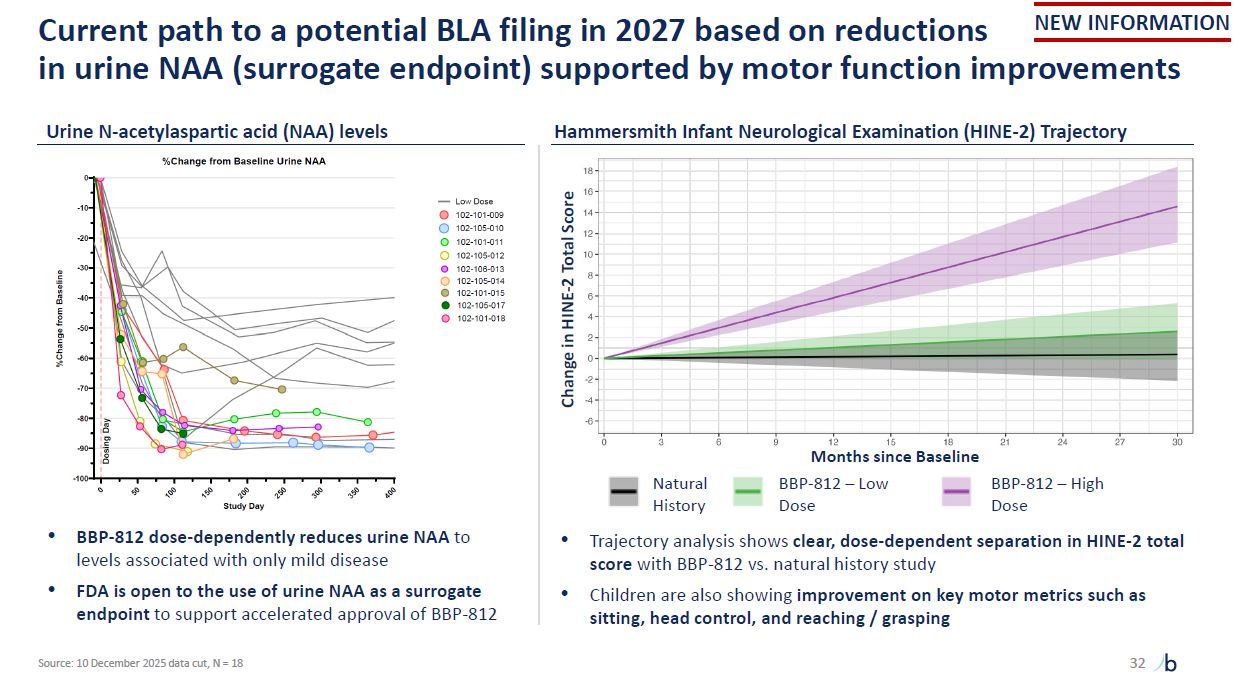
Current path to a potential BLA filing in 2027 based on reductions in urine NAA
(surrogate endpoint) supported by motor function improvements 32 Urine N-acetylaspartic acid (NAA) levels Hammersmith Infant Neurological Examination (HINE-2) Trajectory BBP-812 dose-dependently reduces urine NAA to levels associated with
only mild disease FDA is open to the use of urine NAA as a surrogate endpoint to support accelerated approval of BBP-812 Trajectory analysis shows clear, dose-dependent separation in HINE-2 total score with BBP-812 vs. natural history
study Children are also showing improvement on key motor metrics such as sitting, head control, and reaching / grasping Change in HINE-2 Total Score Natural History BBP-812 – Low Dose BBP-812 – High Dose Months since Baseline NEW
INFORMATION Source: 10 December 2025 data cut, N = 18
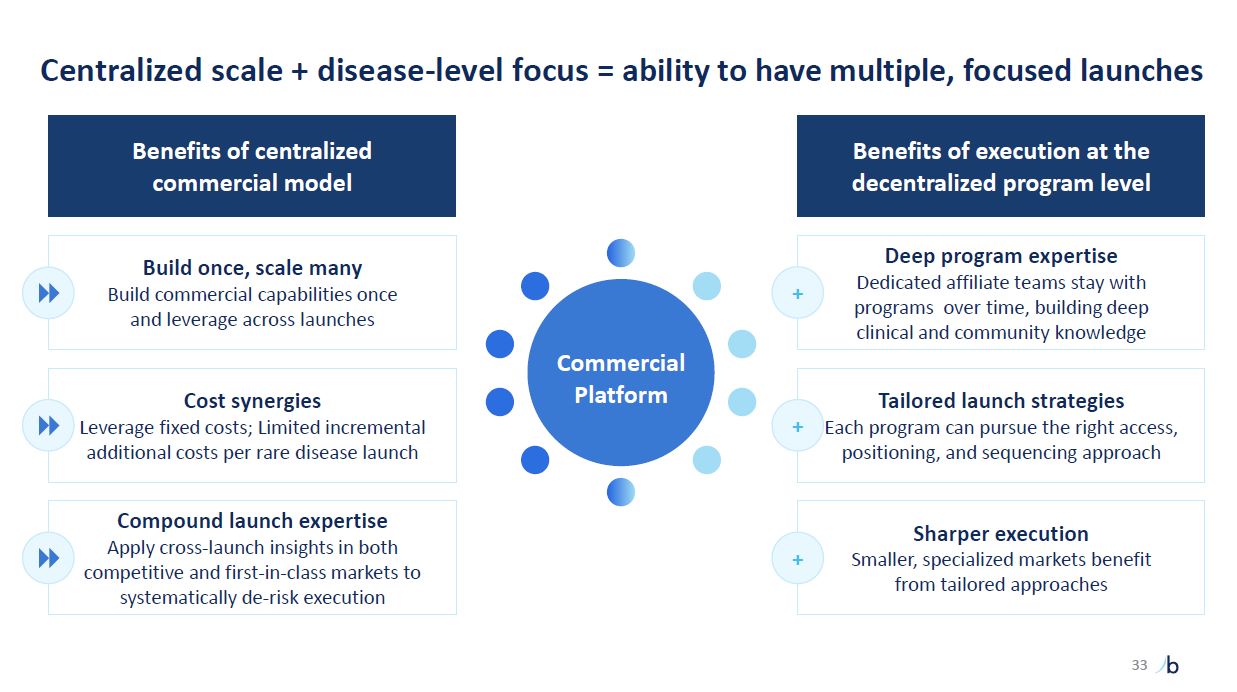
Centralized scale + disease-level focus = ability to have multiple, focused
launches 33 Deep program expertise Dedicated affiliate teams stay with programs over time, building deep clinical and community knowledge Tailored launch strategies Each program can pursue the right access, positioning, and sequencing
approach Sharper execution Smaller, specialized markets benefit from tailored approaches Benefits of execution at the decentralized program level Benefits of centralized commercial model Build once, scale many Build commercial
capabilities once and leverage across launches Cost synergies Leverage fixed costs; Limited incremental additional costs per rare disease launch Compound launch expertise Apply cross-launch insights in both competitive and first-in-class
markets to systematically de-risk execution + + + Commercial Platform

Our future: It’s still day 1 in genetic disease 34

We are at day 1 of genetic medicine 35 Maher Nature 2008; Manolio Nature
2009. “..we show that 12,111 independent SNPs that are significantly associated with height account for nearly all of the common SNP- based heritability.” - Yengo et al, Nature 2022 “We identified 15 traits with no significant difference
between WGS-based and pedigree-based heritability estimates, suggesting their heritability is fully accounted for by WGS data.” - Wainschtein et al, Nature 2025 Today, missing heritability is being explained
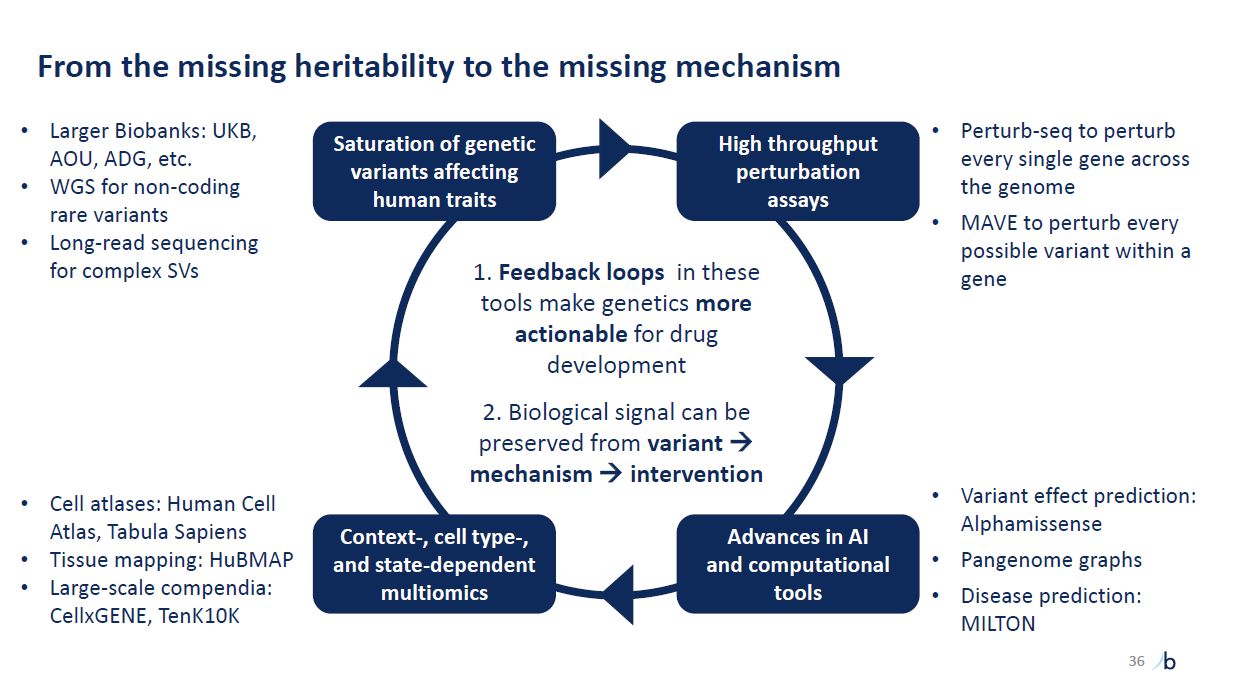
From the missing heritability to the missing mechanism Variant effect prediction:
Alphamissense Pangenome graphs Disease prediction: MILTON 36 Feedback loops in these tools make genetics more actionable for drug development Biological signal can be preserved from variant mechanism intervention High throughput
perturbation assays Advances in AI and computational tools Context-, cell type-, and state-dependent multiomics Saturation of genetic variants affecting human traits Larger Biobanks: UKB, AOU, ADG, etc. WGS for non-coding rare
variants Long-read sequencing for complex SVs Cell atlases: Human Cell Atlas, Tabula Sapiens Tissue mapping: HuBMAP Large-scale compendia: CellxGENE, TenK10K Perturb-seq to perturb every single gene across the genome MAVE to perturb every
possible variant within a gene
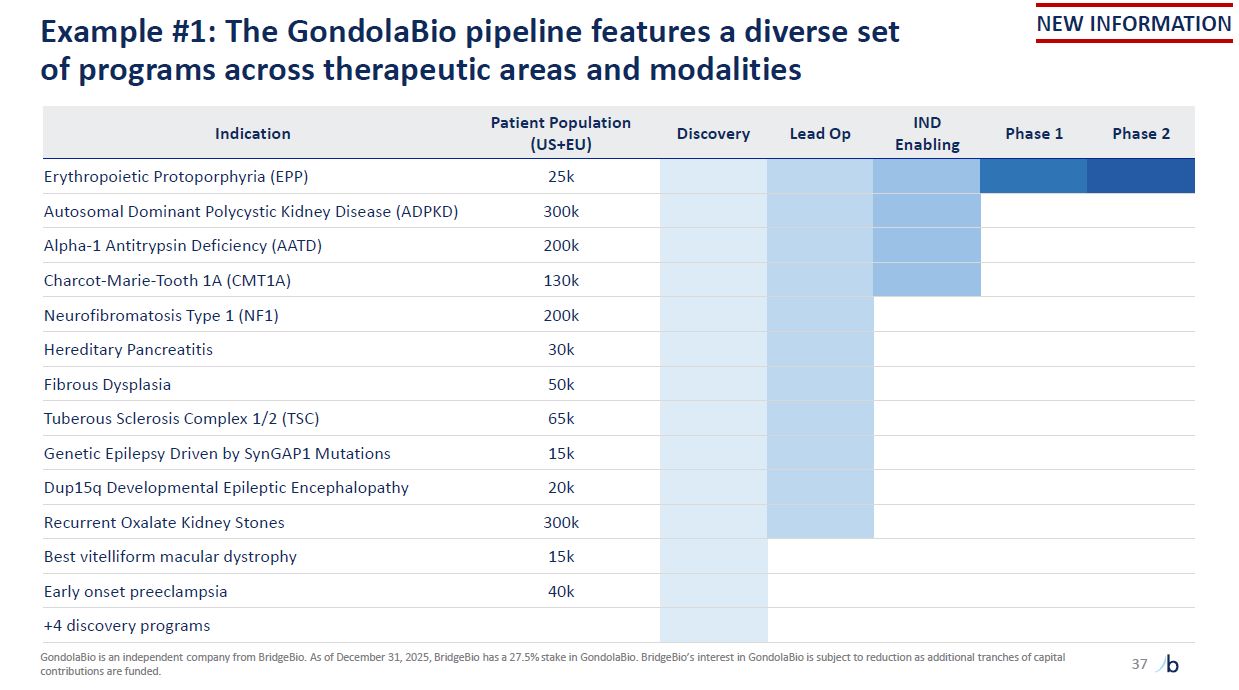
Example #1: The GondolaBio pipeline features a diverse set of programs across
therapeutic areas and modalities 37 GondolaBio is an independent company from BridgeBio. As of December 31, 2025, BridgeBio has a 27.5% stake in GondolaBio. BridgeBio’s interest in GondolaBio is subject to reduction as additional tranches of
capital contributions are funded. NEW INFORMATION Indication Patient Population (US+EU) Discovery Lead Op IND Enabling Phase 1 Phase 2 Erythropoietic Protoporphyria (EPP) 25k Autosomal Dominant Polycystic Kidney Disease
(ADPKD) 300k Alpha-1 Antitrypsin Deficiency (AATD) 200k Charcot-Marie-Tooth 1A (CMT1A) 130k Neurofibromatosis Type 1 (NF1) 200k Hereditary Pancreatitis 30k Fibrous Dysplasia 50k Tuberous Sclerosis Complex 1/2 (TSC) 65k Genetic
Epilepsy Driven by SynGAP1 Mutations 15k Dup15q Developmental Epileptic Encephalopathy 20k Recurrent Oxalate Kidney Stones 300k Best vitelliform macular dystrophy 15k Early onset preeclampsia 40k +4 discovery programs
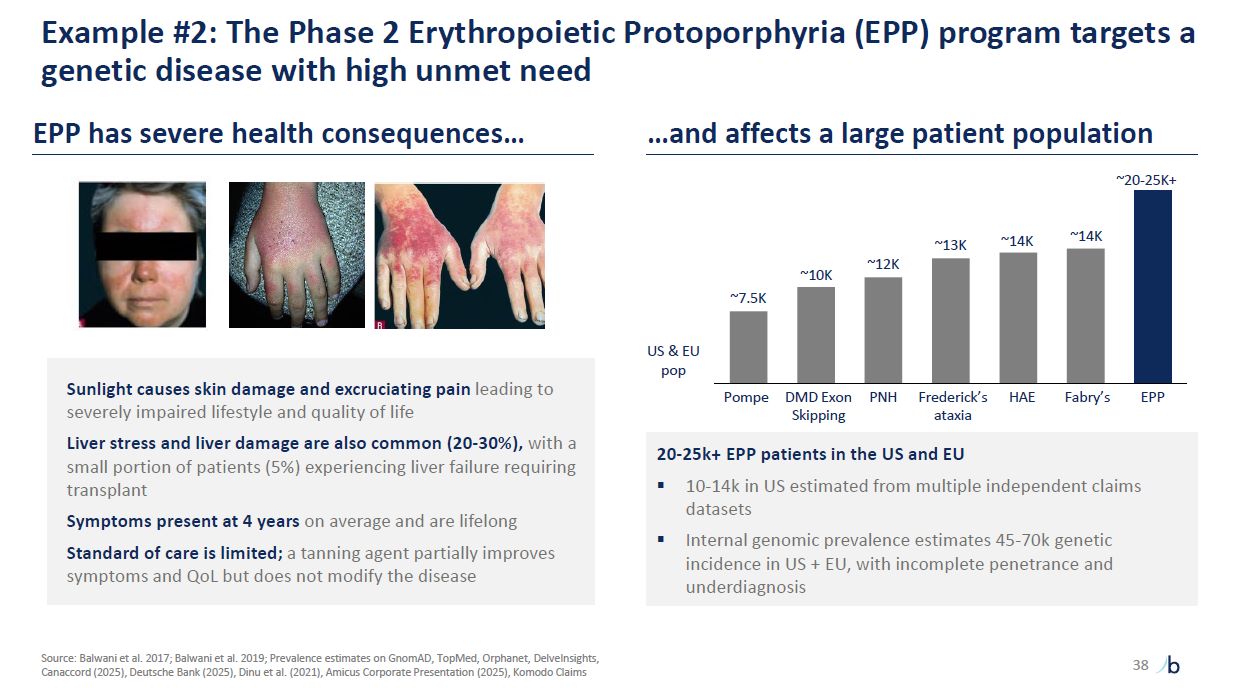
Example #2: The Phase 2 Erythropoietic Protoporphyria (EPP) program targets a
genetic disease with high unmet need Sunlight causes skin damage and excruciating pain leading to severely impaired lifestyle and quality of life Liver stress and liver damage are also common (20-30%), with a small portion of patients (5%)
experiencing liver failure requiring transplant Symptoms present at 4 years on average and are lifelong Standard of care is limited; a tanning agent partially improves symptoms and QoL but does not modify the disease EPP has severe health
consequences… …and affects a large patient population 20-25k+ EPP patients in the US and EU 10-14k in US estimated from multiple independent claims datasets Internal genomic prevalence estimates 45-70k genetic incidence in US + EU, with
incomplete penetrance and underdiagnosis Pompe DMD Exon PNH Skipping Frederick’s HAE Fabry’s EPP ataxia US & EU pop ~7.5K ~10K ~12K ~13K ~14K ~20-25K+ ~14K Source: Balwani et al. 2017; Balwani et al. 2019; Prevalence estimates
on GnomAD, TopMed, Orphanet, DelveInsights, Canaccord (2025), Deutsche Bank (2025), Dinu et al. (2021), Amicus Corporate Presentation (2025), Komodo Claims 38
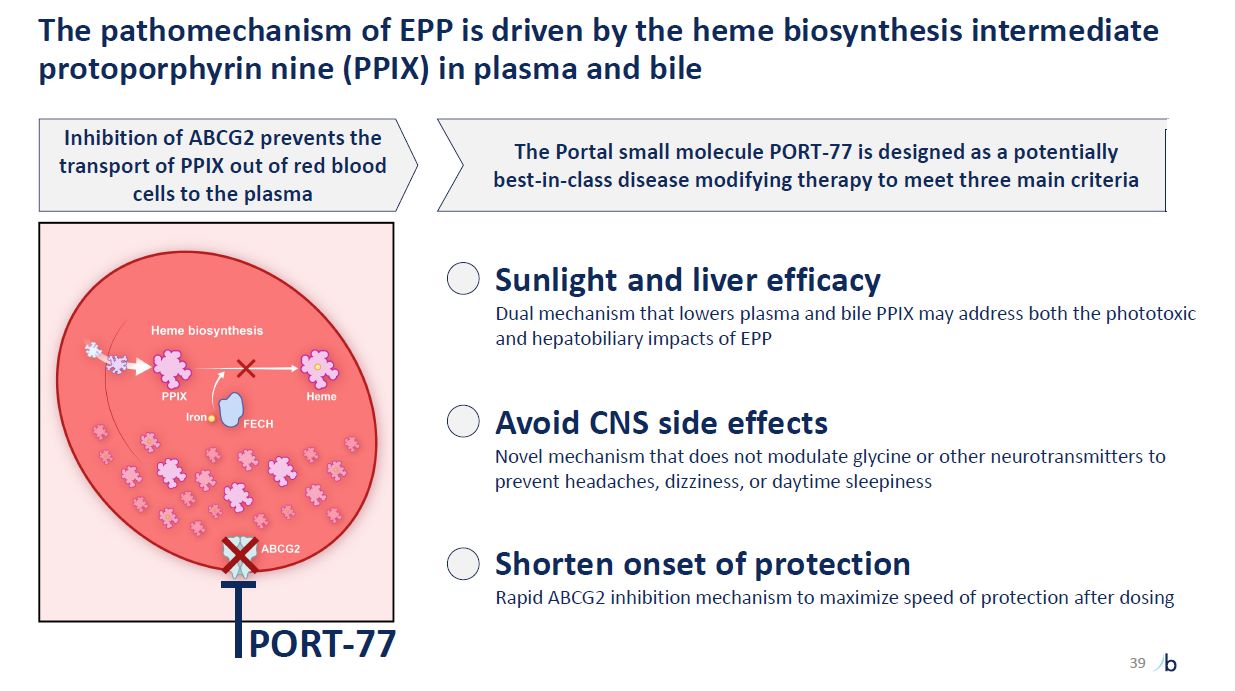
The pathomechanism of EPP is driven by the heme biosynthesis intermediate
protoporphyrin nine (PPIX) in plasma and bile Inhibition of ABCG2 prevents the transport of PPIX out of red blood cells to the plasma Sunlight and liver efficacy Dual mechanism that lowers plasma and bile PPIX may address both the phototoxic
and hepatobiliary impacts of EPP Avoid CNS side effects Novel mechanism that does not modulate glycine or other neurotransmitters to prevent headaches, dizziness, or daytime sleepiness Shorten onset of protection Rapid ABCG2 inhibition
mechanism to maximize speed of protection after dosing PORT-77 The Portal small molecule PORT-77 is designed as a potentially best-in-class disease modifying therapy to meet three main criteria 39

PORT-77 targets EPP at its source by preventing transport of PPIX out of red blood
cells into the plasma, skin, and bile Source: Internal Data 40 PPIX RBCs Plasma, skin, bile ABCG2 PORT-77 has a potential best-in-disease drug profile Mechanism PPIX RBCs PPIX Plasma, skin, bile ABCG2 inhibition keeps PPIX out of
the plasma, skin, and bile ABCG2 inhibition PORT-77 is a small molecule ABCG2 inhibitor Orally bioavailable 15nM IC50 for ABCG2 inhibition Highly selective for ABCG2 over other transporters Human half-life (t1/2): 10-21 hrs Favorable
ADME properties Composition of matter IP through at least 2044 Which addresses all aspects of EPP safely and rapidly Addresses both skin and liver symptoms Highly safe and well-tolerated in preclinical + phase 1 dosing No CNS side
effects Onset of action in minutes vs weeks
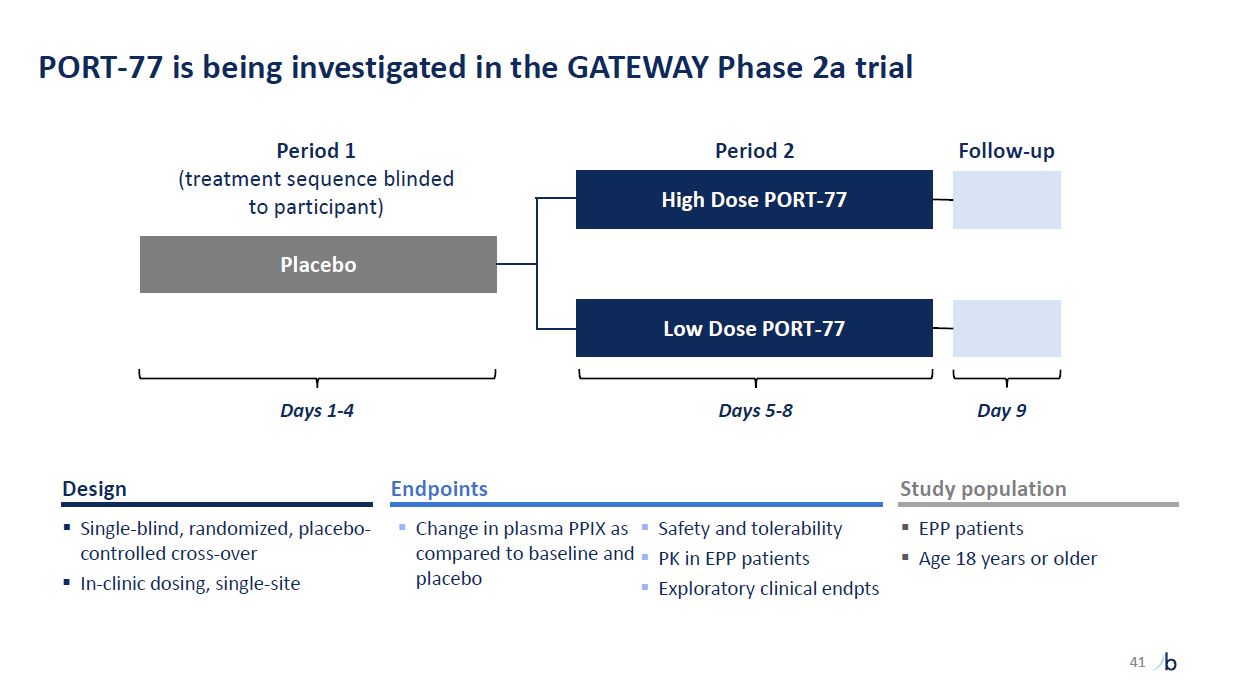
PORT-77 is being investigated in the GATEWAY Phase 2a trial Placebo High Dose
PORT-77 Period 1 (treatment sequence blinded to participant) Period 2 Follow-up Low Dose PORT-77 placebo Exploratory clinical endpts Endpoints Change in plasma PPIX as Safety and tolerability compared to baseline and PK in EPP
patients Single-blind, randomized, placebo-controlled cross-over In-clinic dosing, single-site Design Study population EPP patients Age 18 years or older Days 1-4 Days 5-8 Day 9 41
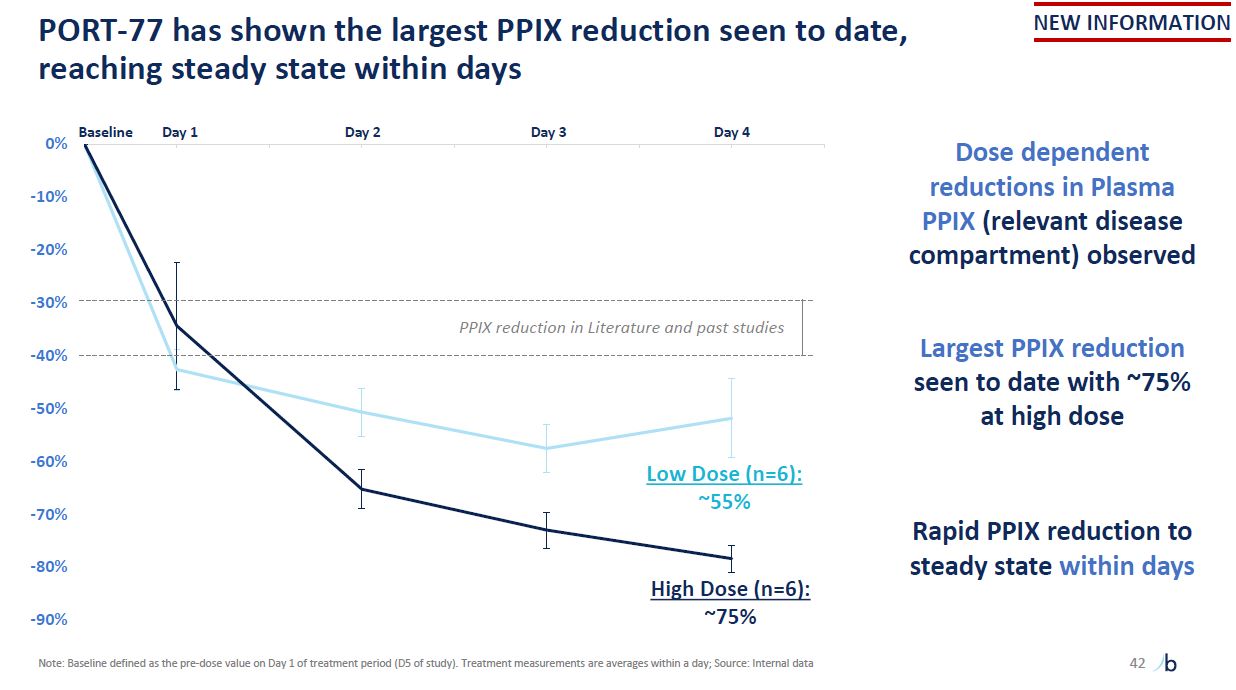
-90% -80% -70% -60% -50% -40% -30% -20% -10% 0% Baseline PORT-77 has
shown the largest PPIX reduction seen to date, reaching steady state within days Low Dose (n=6): ~55% High Dose (n=6): ~75% Dose dependent reductions in Plasma PPIX (relevant disease compartment) observed Largest PPIX reduction seen to
date with ~75% at high dose Rapid PPIX reduction to steady state within days PPIX reduction in Literature and past studies Day 1 Day 2 Day 3 Day 4 42 NEW INFORMATION Note: Baseline defined as the pre-dose value on Day 1 of treatment
period (D5 of study). Treatment measurements are averages within a day; Source: Internal data
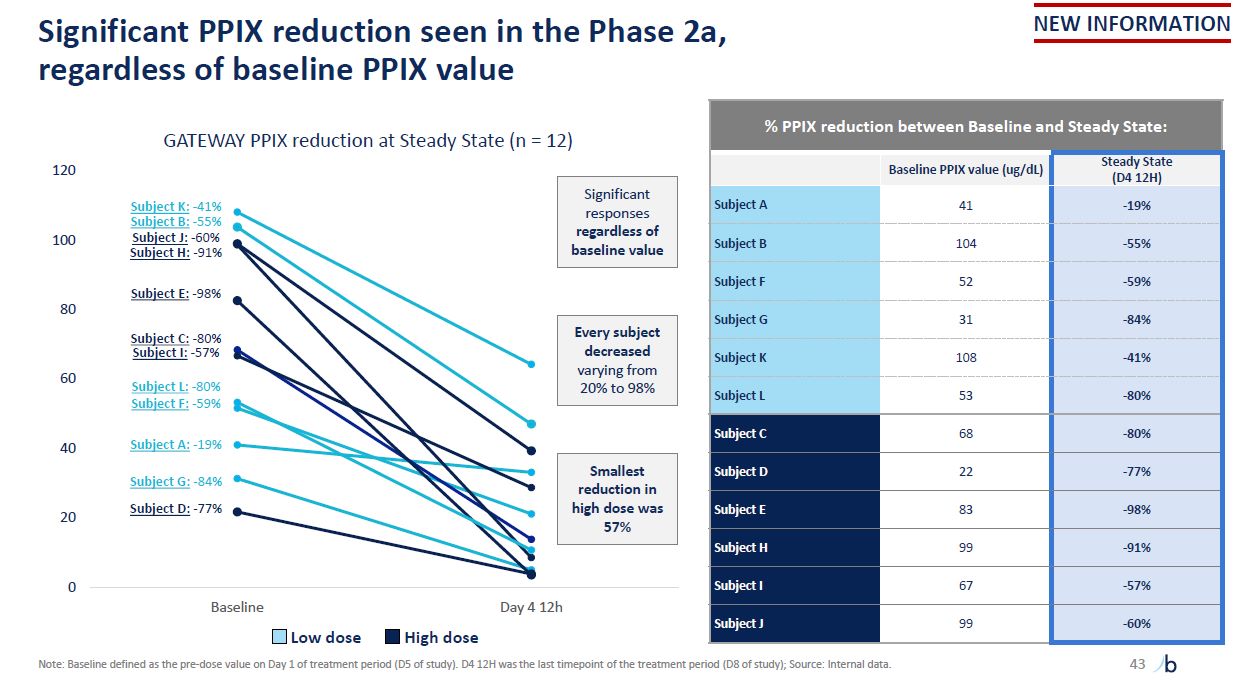
Significant PPIX reduction seen in the Phase 2a, regardless of baseline PPIX
value 0 20 40 60 Subject E: -98% 80 100 GATEWAY PPIX reduction at Steady State (n = 12) 120 Baseline Day 4 12h Subject K: -41% Subject B: -55% Subject J: -60% Subject H: -91% Subject A: -19% Subject C: -80% Subject I:
-57% Subject D: -77% Subject G: -84% Subject L: -80% Subject F: -59% % PPIX reduction between Baseline and Steady State: Baseline PPIX value (ug/dL) Steady State (D4 12H) Subject A 41 -19% Subject B 104 -55% Subject
F 52 -59% Subject G 31 -84% Subject K 108 -41% Subject L 53 -80% Subject C 68 -80% Subject D 22 -77% Subject E 83 -98% Subject H 99 -91% Subject I 67 -57% Subject J 99 -60% Low dose High dose Note: Baseline
defined as the pre-dose value on Day 1 of treatment period (D5 of study). D4 12H was the last timepoint of the treatment period (D8 of study); Source: Internal data. Significant responses regardless of baseline value Every subject decreased
varying from 20% to 98% Smallest reduction in high dose was 57% NEW INFORMATION 43
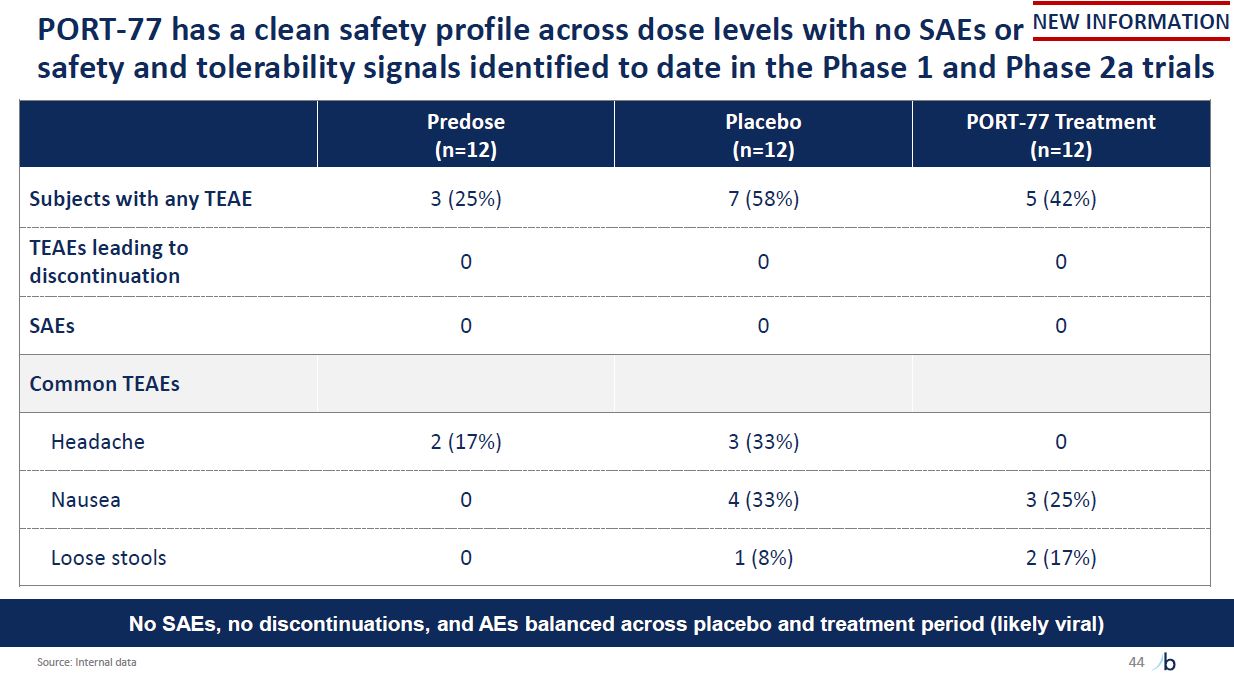
safety and tolerability signals identified to date in the Phase 1 and Phase 2a
trials PORT-77 has a clean safety profile across dose levels with no SAEs or NEW INFORMATION Predose (n=12) Placebo (n=12) PORT-77 Treatment (n=12) Subjects with any TEAE 3 (25%) 7 (58%) 5 (42%) TEAEs leading to
discontinuation 0 0 0 SAEs 0 0 0 Common TEAEs Headache 2 (17%) 3 (33%) 0 Nausea 0 4 (33%) 3 (25%) Loose stools 0 1 (8%) 2 (17%) No SAEs, no discontinuations, and AEs balanced across placebo and treatment period (likely
viral) Source: Internal data 44

The EPP program has achieved proof-of-concept for a potential best-in-disease
profile Consistent and potential best-in-class PPIX reduction; Dual-mechanism independently treating sunlight and liver symptoms Clean safety profile with no notable AEs in treatment group and lack of CNS effects to date PPIX reduction seen
within hours, days to steady-state effect 45
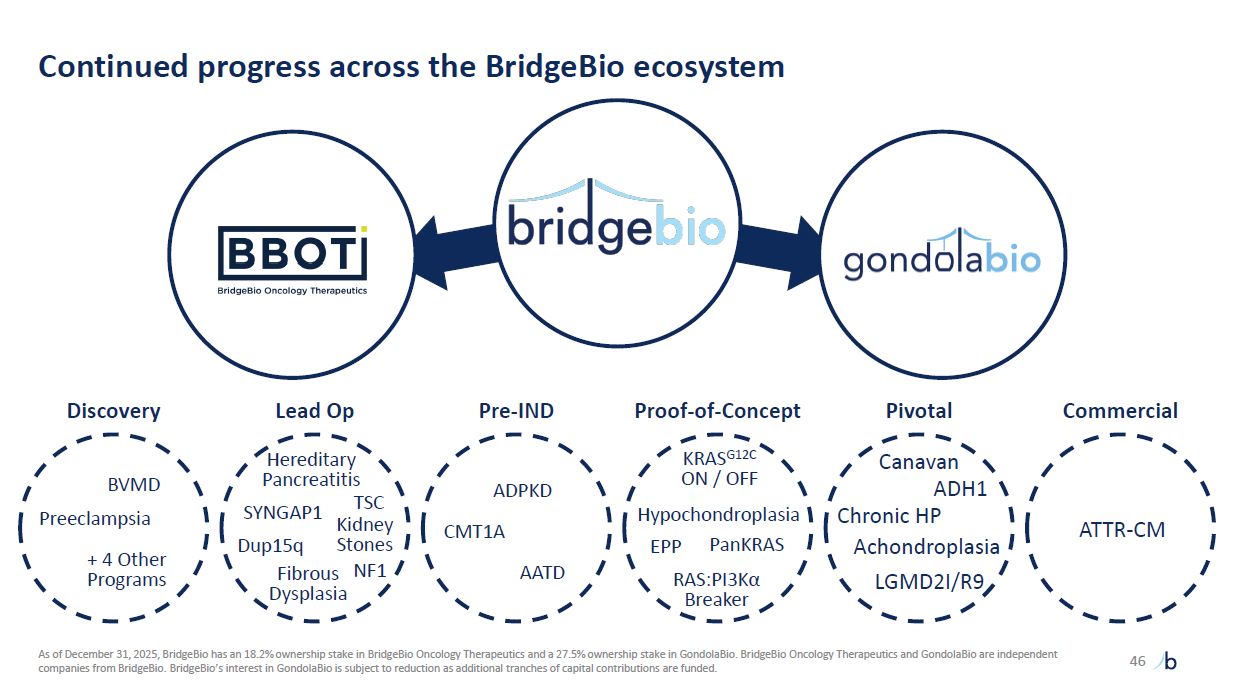
Continued progress across the BridgeBio ecosystem 46 + 4 Other
Programs BVMD Preeclampsia Fibrous Dysplasia SYNGAP1 Dup15q Kidney Stones ADPKD AATD CMT1A RAS:PI3Kα Breaker PanKRAS KRASG12C ON / OFF HHypypoochchoonndropElPaPsia Canavan ADH1 Chronic
HP Achondroplasia LGMD2I/R9 ATTR-CM Pivotal Discovery Lead Op Pre-IND Proof-of-Concept Commercial As of December 31, 2025, BridgeBio has an 18.2% ownership stake in BridgeBio Oncology Therapeutics and a 27.5% ownership stake in
GondolaBio. BridgeBio Oncology Therapeutics and GondolaBio are independent companies from BridgeBio. BridgeBio’s interest in GondolaBio is subject to reduction as additional tranches of capital contributions are funded. EPP NF1 Hereditary
Pancreatitis TSC

47 Key takeaways today – significant momentum across the portfolio NEW
INFORMATION Attruby: $146M1 in Q4 net product revenue; 6,629 unique patient prescriptions; and >25% NBRx share New antibody depleter program announced for ATTR-CM ATTR-CM LPLV for Phase 3 achondroplasia trial achieved LPI for Phase 2
hypochondroplasia trial achieved Infigratinib Broad benefit of BBP-418 in all subgroups across α-controlled efficacy endpoints at 12 months Highly clinically meaningful and stat sig. 2.6 point benefit on NSAD relative to placebo at 12
months Recommendation from FDA to orient NDA toward traditional approval BBP-418 Additional data demonstrating dose-dependent reductions in urine NAA and motor function improvements Current path to potential BLA filing in
2027 BBP-812 Rapid uptake in diagnosis of ADH1 with >1,700 unique patients identified in claims since October 2023 FDA alignment on and path forward with Phase 3 RECLAIM-HP trial in Chronic HP; expected to initiate in summer
2026 Encaleret Positive Phase 2a data for PORT-77 in EPP GondolaBio 1Represents preliminary, unaudited results for the fourth quarter ended December 31, 2025, based on management’s current expectations and subject to completion of year end
audit procedures. See Forward Looking Statements and Disclaimer on slide 2 regarding risks and uncertainties that could cause actual results to differ. Note: Unique patient prescriptions and NBRx share as of 12/31/2025. GondolaBio is an
independent company from BridgeBio. As of December 31, 2025, BridgeBio has a 27.5% stake in GondolaBio. BridgeBio’s interest in GondolaBio is subject to reduction as additional tranches of capital contributions are funded. NSAD=North Star
Assessment for Girdle Type Muscular Dystrophies. EPP = Erythropoietic Protoporphyria

Cash burn declined in Q4 2025 relative to Q3 2025, driven by rising revenues and
improving operating leverage BridgeBio ended 2025 with $587.5M1 in cash, cash equivalents, and marketable securities We are well-financed to hit a drumbeat of potential milestones in 2026 and beyond 1Represents preliminary, unaudited results
as of December 31, 2025, subject to completion of year-end audit procedures. See Forward Looking Statements and Disclaimer on slide 2 regarding risks and uncertainties that could cause actual results to differ. ACH=achondroplasia.
HCH=hypochondroplasia. CHP=chronic hypoparathyroidism. Dates reflect anticipated timing. 48 1H 2026 Infigratinib: ACH Topline Encaleret: Initiate P2/3 pediatric ADH1 Encaleret: NDA filing BBP-418: NDA filing Encaleret: Initiate P3 CHP
trial 2H 2026 Infigratinib: HCH P2 data readout BBP-418: FDA approval and product launch 1H 2027 Encaleret: FDA approval and product launch NEW INFORMATION