BRIGHT MINDS BIOSCIENCES INC.
![]()
ANNUAL INFORMATION FORM
For the Fiscal Year Ended September 30, 2025
Dated December 23, 2025
|
INTRODUCTORY NOTES |
3 |
|
Introduction |
3 |
|
Date of Information |
3 |
|
Currency and Exchange Rates |
3 |
|
Use of Market and Industry Data |
3 |
|
FORWARD-LOOKING INFORMATION |
3 |
|
GLOSSARY OF TERMS |
8 |
|
CORPORATE STRUCTURE |
11 |
|
Name, Address and Incorporation |
11 |
|
Intercorporate Relationships |
11 |
|
GENERAL DEVELOPMENT OF THE BUSINESS |
11 |
|
Overview |
11 |
|
Three-Year History |
12 |
|
DESCRIPTION OF THE BUSINESS |
14 |
|
Company Overview |
14 |
|
Principal Products and Services |
14 |
|
Production and Services |
17 |
|
Specialized Skill and Knowledge |
17 |
|
Competitive Conditions |
17 |
|
Intangible Properties |
18 |
|
Trademarks |
25 |
|
Web Domains |
25 |
|
Government Regulation |
25 |
|
Cycles |
30 |
|
Economic Dependence |
30 |
|
Changes to Contracts |
30 |
|
Environmental Protection |
30 |
|
Employees |
30 |
|
Foreign Operations |
30 |
|
Lending |
31 |
|
Bankruptcy and Similar Procedures |
31 |
|
Reorganizations |
31 |
|
Social or Environmental Policies |
31 |
|
RISK FACTORS |
32 |
|
Risks Related to the Business of the Company |
32 |
|
Risks Related to Our Common Shares |
48 |
|
DIVIDENDS AND DISTRIBUTIONS |
52 |
|
DESCRIPTION OF CAPITAL STRUCTURE |
52 |
|
Common Shares |
52 |
|
Warrants |
52 |
|
Options |
53 |
|
MARKET FOR SECURITIES |
53 |
|
Trading Price and Volume |
53 |
|
Prior Sales |
55 |
|
ESCROWED SECURITIES AND SECURITIES SUBJECT TO CONTRACTUAL RESTRICTIONS ON TRANSFER |
55 |
|
DIRECTORS AND OFFICERS |
55 |
|
Name, Occupation and Security Holding |
55 |
|
Term of Office |
56 |
|
Director and Officer Share Ownership |
56 |
|
Biographies |
56 |
|
Cease Trade Orders, Bankruptcies, Penalties or Sanctions |
59 |
|
Conflicts of Interest |
60 |
|
LEGAL PROCEEDINGS AND REGULATORY ACTIONS |
60 |
|
PROMOTERS |
60 |
|
INTERESTS OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS |
60 |
|
AUDITOR, TRANSFER AGENT AND REGISTRAR |
61 |
|
Auditor |
61 |
|
Transfer Agent and Registrar |
61 |
|
MATERIAL CONTRACTS |
61 |
|
AUDIT COMMITTEE |
61 |
|
Audit Committee Charter |
61 |
|
Composition of the Audit Committee |
61 |
|
Relevant Education and Experience |
62 |
|
Audit Committee Oversight |
62 |
|
Pre-Approval Policies and Procedures |
62 |
|
External Auditor Service Fees |
63 |
|
INTERESTS OF EXPERTS |
63 |
|
Names of Experts |
63 |
|
Interests of Experts |
63 |
|
ADDITIONAL INFORMATION |
63 |
|
SCHEDULE A |
A-1 |
INTRODUCTORY NOTES
Introduction
This Annual Information Form (“AIF”) has been prepared in accordance with Form 51-102F2 – Annual Information Form and should be read in conjunction with Bright Minds Biosciences Inc.’s (the “Company” or “Bright Minds”) audited consolidated financial statements (and accompanying notes) for the year ended September 30, 2025, and Management’s Discussion and Analysis for the year ended September 30, 2025. These documents, along with additional information about the Company are available on its issuer profile on SEDAR+ at www.sedarplus.ca.
For an explanation of the capitalized terms and expressions and certain defined terms, please refer to the “GLOSSARY OF TERMS” below.
Date of Information
All information contained in this AIF is as of September 30, 2025, with subsequent events disclosed to December 23, 2025, unless otherwise indicated.
Currency and Exchange Rates
All dollar amounts herein are expressed in Canadian dollars unless otherwise indicated.
Use of Market and Industry Data
This AIF includes market and industry data that has been obtained from third party sources, including industry publications, as well as industry data prepared by the Company’s management on the bases of its knowledge of and experience in the industry in which the Company operates (including management’s estimates and assumptions relating to the industry based on that knowledge). Management’s knowledge of the industry has been developed through its experience and lengthy participation in the industry. Management believes that its industry data is accurate and that its estimates and assumptions are reasonable, but there is no assurance as to the accuracy or completeness of this data. Third party sources generally state that the information contained therein has been obtained from sources believed to be reliable, but there is no assurance as to the accuracy or completeness of included information. Although the Company’s management believes it to be reliable, it has not independently verified any of the data from third party sources referred to in this AIF or ascertained the underlying economic assumptions relied upon by such sources.
FORWARD-LOOKING INFORMATION
This AIF contains certain statements, which may constitute “forward-looking information” within the meaning of Canadian securities law requirements. These forward-looking statements are made as of the date of this AIF and the Company does not intend, and does not assume any obligation, to update these forward-looking statements, except as required under applicable securities legislation. Forward-looking statements relate to future events or future performance and reflect Company management’s expectations or beliefs regarding future events. In certain cases, forward-looking statements can be identified by the use of words such as “plans”, “expects” or “does not expect”, “is expected”, “budget”, “scheduled”, “estimates”, “forecasts”, “pipeline”, “intends”, “anticipates” or “does not anticipate”, or “believes”, or variations of such words and phrases or statements that certain actions, events or results “may”, “could”, “would”, “might” or “will be taken”, “occur” or “be achieved” or the negative of these terms or comparable terminology. In this document, certain forward- looking statements are identified by words including “may”, “future”, “expected”, “intends” and “estimates”. By their very nature, forward-looking statements involve known and unknown risks, uncertainties and other factors, which may cause the actual results, performance or achievements of the Company to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. The Company provides no assurance that forward-looking statements will prove to be accurate, as actual results and future events could differ materially from those anticipated in such statements. Accordingly, readers should not place undue reliance on forward-looking statements. Certain forward-looking statements in this AIF include, but are not limited to the following:
|
● |
the Company’s expectations regarding the achievement of clinical and regulatory milestones; |
|
● |
the Company’s expectations regarding its revenue, expenses and research and development operations; |
|
● |
the Company’s anticipated cash needs and its needs for additional financing; |
|
● |
the Company’s intention to grow the business and its operations; |
|
● |
expectations with respect to the success of its research and development of serotonergic therapeutics; |
|
● |
expectations regarding growth rates, growth plans and strategies of the Company; |
|
● |
expectations that provisional patent applications will be converted to regular patent applications or refiled as new provisional patent applications 12 months from their filing dates; |
|
● |
expectations that prosecution of patent applications that have entered the national/regional phase will begin; |
|
● |
the Company’s strategy with respect to the expansion and protection of its intellectual property; |
|
● |
the medical benefits, safety, efficacy, dosing and consumer acceptance of serotonergic therapeutics; |
|
● |
the Company’s ability to comply with provincial, federal, local and regulatory agencies in the United States, Canada and other jurisdictions in which the Company operates; |
|
● |
the Company’s competitive position and the regulatory environment in which the Company operates; |
|
● |
the Company’s expected business objectives for the next 12 months; |
|
● |
the Company’s plans with respect to the payment of dividends; |
|
● |
beliefs and intentions regarding the ownership of material trademarks and domain names used in connection with the design, production, marketing, distribution and sale of the Company’s products and services; |
|
● |
the Company’s ability to obtain additional funds through the sale of equity or debt commitments; |
|
● |
the Company’s ability to obtain the necessary regulatory approvals; |
|
● |
expectations that regulatory requirements will be maintained; |
|
● |
expectations related to general business and economic conditions; |
|
● |
the Company’s ability to successfully execute its plans and intentions; |
|
● |
the availability of financing on reasonable terms to the Company; |
|
● |
the Company’s ability to attract and retain skilled staff; |
|
● |
expectations about market competition; |
|
● |
expectations about the products, services and technology offered by the Company’s competitors; and |
|
● |
expectations that the Company’s current good relationships with its suppliers, service providers and other third parties will be maintained. |
The above and other aspects of the Company’s anticipated future operations are forward-looking in nature and, as a result, are subject to certain risks and uncertainties. Although the Company believes that the expectations reflected in these forward-looking statements are reasonable, undue reliance should not be placed on them as actual results may differ materially from the forward-looking statements. Such forward-looking statements are estimates reflecting the Company’s best judgment based upon current information and involve a number of risks and uncertainties, and there can be no assurance that other factors will not affect the accuracy of such forward-looking statements. Such factors include but are not limited to:
|
● |
the Company’s limited operating history; |
|
● |
the Company’s actual financial position and results of operations may differ materially from the expectations of the Company’s management; |
|
● |
the Company may not be successful in its efforts to identify, license or discover additional product candidates; |
|
● |
the Company may be required to obtain and maintain certain permits, licenses, and approvals in the jurisdictions where its products or technologies are being researched, developed, or commercialized; |
|
● |
the Company may encounter substantial delays or difficulties with its current and future clinical trials; |
|
● |
clinical trials are very expensive, time consuming and difficult to design and implement; |
|
● |
the Company’s current and future clinical trials or those of its current or future collaborators may reveal significant adverse events not seen in pre-clinical and non-clinical studies and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of the Company’s product candidates; |
|
● |
the Company has limited experience in completing clinical trials and has only completed one phase one drug trial to date; |
|
● |
if the Company experience delays or difficulties in the enrolment of patients in clinical trials, receipt of regulatory approvals could be delayed or prevented; |
|
● |
success in pre-clinical studies or clinical trials may not be predictive of results in future clinical trials; |
|
● |
interim, “topline,” and preliminary data from the Company’s clinical trials that the Company announces or publishes from time to time may change as more patient data becomes available and are subject to audit and verification procedures that could result in material changes in the final data; |
|
● |
the Company may not be successful in its efforts to identify, license or discover additional product candidates; |
|
● |
there is no assurance that the Company will turn a profit or generate immediate revenues; |
|
● |
the Company’s intellectual property and licenses thereto; |
|
● |
the Company may not achieve the timelines for project development set out in this AIF; |
|
● |
the Company may need additional capital for future operations; |
|
● |
the Company faces product liability exposure; |
|
● |
there may be health and safety issues related to the Company’s products; |
|
● |
the Company has international operations, which subject the Company to risks inherent with operations outside of Canada; |
|
● |
the Company may be adversely impacted by changes in U.S. and international trade policies; |
|
● |
the Company, its investors and its securities may be adversely impacted by changes in U.S. laws; |
|
● |
exchange rate fluctuations between the U.S. dollar and the Canadian dollar; |
|
● |
changes to patent laws or the interpretation of patent laws; |
|
● |
the Company may not be able to enforce its intellectual property rights throughout the world; |
|
● |
the lack of product for commercialization; |
|
● |
the failure to develop new and innovative products; |
|
● |
the lack of experience of the Company/management in marketing, selling, and distribution products; |
|
● |
the size of the Company’s target market is difficult to quantify; |
|
● |
ongoing sales of Common Shares will dilute current shareholders; |
|
● |
clinical and pre-clinical drug development is lengthy, costly and carries uncertain outcomes; |
|
● |
potentials for conflicts of interest for the Company’s officers and directors; |
|
● |
in certain circumstances, the Company’s reputation could be damaged; |
|
● |
negative operating cash flow; |
|
● |
forward-looking statements may prove to be inaccurate; |
|
● |
the potential for a material weakness in the Company’s internal controls over financial reporting; |
|
● |
difficulties with forecasts; |
|
● |
the Company’s executive officers and directors exercise a significant level of control; |
|
● |
market price of Common Shares and volatility; |
|
● |
the Company does not intend to pay dividends; |
|
● |
the ability to buy and sell Common Shares may be limited by FINRA’s sales practice requirements; |
|
● |
the Company is a foreign private issuer within the meaning of the rules under the Exchange Act and is exempt from certain disclosure provisions; |
|
● |
as an “emerging growth company”, the Company is subject to lessened disclosure requirements under U.S. securities laws; |
|
● |
costs of maintaining status as a public company; and |
|
● |
other risks as set out under “Risk Factors” below. |
[Remainder of Page Intentionally Left Blank]
GLOSSARY OF TERMS
The following is a glossary of certain terms used in this AIF:
“Agents” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS – Three-Year History – Financial Year Ended September 30, 2025”.
“AIF” means this annual information form of the Company dated December 23, 2025 for the year ended September 30, 2025.
“ATM” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS – Three-Year History – Financial Year Ended September 30, 2025”.
“Audit Committee” means a committee established by and among the Board for the purpose of assisting the Board in fulfilling its financial oversight responsibilities.
“Bankruptcy Code” means the Bankruptcy Code of the United States or any successor statute.
“Base Shelf Registration Statement” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2025”.
“BCBCA” means the Business Corporations Act (British Columbia).
“BCSC” means the British Columbia Securities Commission.
“BLA” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Government Regulation– United States Government Regulation.”
“Board of Directors” or “Board” means the board of directors of the Company.
“Code of Ethics” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – “Social or Environmental Policies”.
“Common Shares” means common shares in the capital of the Company.
“Company” or “Bright Minds Biosciences” means Bright Minds Biosciences Inc.
“Complete Response Letter” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation – The FDA’s Decision on an NDA or BLA.”
“CSE” means the Canadian Securities Exchange.
“December 2022 Unit Offering” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2023”.
“December 2023 Private Placement” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2024”.
“DMG” means DMG Blockchain Solutions Inc.
“Equity Distribution Agreement” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2025”.
“Exchange Act” means the United States Exchange Act of 1934.
“Exclusive License Agreement” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Intangible Properties- Patents and Patent Applications– Kozikowski-Roth Patents”.
“FDCA” means the United States Food, Drug and Cosmetics Act.
“FDA” means the United States Food and Drug Administration.
“FINRA” means the Financial Industry Regulation Authority.
“GCP” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation–United States Government Regulation.”
“IFRS” means International Financial Reporting Standards.
“IND” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation– United States Government Regulation.”
“Inventions” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Intangible Properties- Patents and Patent Applications– Kozikowski-Roth Patents”.
“IRB” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation– United States Government Regulation.”
“JOBS Act” means the United States Jumpstart our Business Startups (JOBS) Act of 2012.
“License” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Intangible Properties- Patents and Patent Applications– Kozikowski-Roth Patents”.
“NASDAQ” means the Nasdaq Capital Market.
“NI 52-110” has the meaning ascribed thereto under “AUDIT COMMITTEE”.
“November 2024 Private Placement” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2025”.
“Options” means a stock option of the Company.
“PFWs” has the meaning ascribed thereto under “GENERAL DEVELOPMENT OF THE BUSINESS– Three-Year History– Financial Year Ended September 30, 2023”.
“Exclusive License Agreement” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Intangible Properties- Patents and Patent Applications– Kozikowski-Roth Patents”.
“NDA” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation– United States Government Regulation.”
“NDS” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation– Canadian Government Regulation.”
“Roth Kozikowski Agreement” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS– Intangible Properties- Patents and Patent Applications– Kozikowski-Roth Patents”.
“RSU” means a restricted share unit of the Company.
“SEC” means the United States Securities and Exchange Commission.
“Shareholders” means the holders of the Common Shares.
“TPD” has the meaning ascribed thereto under “DESCRIPTION OF THE BUSINESS – Government Regulation– Canadian Government Regulation.”
“TSXV” means the TSX Venture Exchange.
“UIC” means the University of Illinois
“Unit” means a unit in the capital of the Company.
“U.S. Securities Act” means the United States Securities Act of 1933.
“Warrant” means a Common Share purchase warrant in the capital of the Company.
“Warrant Share” means a Common Share to be issued upon the exercise of a Warrant.
[Remainder of Page Intentionally Left Blank]
CORPORATE STRUCTURE
Name, Address and Incorporation
The Company was incorporated on May 31, 2019 under the BCBCA as “1210954 B.C. Ltd.” On March 6, 2020, the Company changed its name to “Bright Minds Biosciences Inc.” The Company’s Canadian headquarters is located at 1122 Mainland St #228, Vancouver, BC V6B 5L1. The Company’s US headquarters is located at 400 N Aberdeen St Suite 900, Chicago, IL 60642. The Company also has offices at 19 Vestry Street, New York, NY 10013
The Company’s registered and records office is located at Suite 1500, 1055 West Georgia Street, P.O. Box 11117, Vancouver, BC V6E 4N7. The Company’s telephone number is 604-661-9496 and its website address is www.brightmindsbio.com.
The Company’s Common Shares are listed on the CSE and the NASDAQ under the trading symbol “DRUG”. The Company is a reporting issuer in the provinces of British Columbia, Alberta, Saskatchewan, Manitoba, Ontario, New Brunswick, Nova Scotia, Prince Edward Island, and Newfoundland and Labrador.
Intercorporate Relationships
The Company has two wholly owned subsidiaries. The Company’s corporate structure is set out below:
GENERAL DEVELOPMENT OF THE BUSINESS
Overview
The Company is a biotechnology company developing innovative therapeutics to improve the lives of patients with severe and life-altering neurological and psychiatric disorders. The Bright Minds pipeline includes novel compounds targeting key receptors in the brain to address conditions with high unmet medical need, including epilepsy, depression, and other central nervous system (CNS) disorders. The Company is focused on delivering breakthrough therapies that can transform patients’ lives. The Company has developed a unique platform of highly selective serotonergic agonists exhibiting selectivity at different serotonergic receptors. This has provided a rich portfolio of new chemical entity (NCE) programs within neurology and psychiatry.
See “DESCRIPTION OF THE BUSINESS” below for additional information on the Company.
Three-Year History
Financial Year Ended September 30, 2023
On December 1, 2022, the Company appointed Mark A. Smith, M.D., Ph.D., as Chief Medical Officer.
On December 2, 2022, the Company closed a non-brokered private placement of (i) 133,200 pre-funded warrants of the Company (“PFWs”) at a price of $6.245 per PFW, and (ii) 194,800 Units at a price of $6.25 per Unit (collectively, the “December 2022 Unit Offering”), for aggregate gross proceeds of $2,049,334. Each PFW was exercisable into one Unit at an exercise price of $0.005 per Unit on the date that is the earlier of (a) the date the holder thereof elects to exercise the PFWs and pays the exercise price therefor, and (b) December 2, 2024. Each Unit was comprised of one Common Share and Warrant. Each Warrant issued in the December 2022 Unit Offering entitled the holder thereof to acquire a Warrant Share at a price of $6.75 per Warrant Share until December 2, 2024.
On December 1, 2022, the Company granted 60,000 Options to Mark A. Smith, M.D., Ph.D., the Company’s Chief Medical Officer. The Options are exercisable at an exercise price of $8.25 for a period of five (5) years from the date of grant.
On December 1, 2022, the Company granted an aggregate of 220,000 RSUs to Ian McDonald, the Company’s Chief Executive Officer, and Jan Pedersen, the Company’s Chief Scientific Officer. The RSUs are subject to vesting provisions pursuant to which 25% will vest annually, commencing on the date of grant.
On January 9, 2023, the Company announced the resignation of Dr. Doug Williamson from its Board of Directors.
On February 17, 2023, the Company announced the appointment of David Weiner, MD to its Board of Directors.
On July 14, 2023, the Company completed the consolidation of its Common Shares on the basis of five (5) pre-consolidation Common Shares to one (1) post-consolidation Common Share.
On August 8, 2023, the Company announced positives results of the qEEG (Quantitative Electroencephalogram) data in Cohort 4 of its first-in-human Phase 1 study of its lead compound, BMB-101.
Financial Year Ended September 30, 2024
On December 22, 2023, the Company closed a fully management-subscribed, non-brokered private placement of 661,765 Units at a price of $1.36 per Unit for aggregate gross proceeds of $900,000 (the “December 2023 Private Placement”). Each Unit consisted of one Common Share and one Warrant. Each Warrant issued in the December 2023 Private Placement grants the holder the right to acquire one Warrant Share at a price of $1.70 per Warrant Share until December 22, 2028. Ian McDonald, the CEO and a director of the Company, was the sole subscriber in the December 2023 Private Placement.
On March 27, 2024, the Company announced the grant of 130,000 Options to three directors and an employee of the Company. The Options are exercisable at an exercise price of $1.84 for a period of five (5) years from the date of grant.
On September 12, 2024, the Company announced the initiation of the BREAKTHROUGH Study, an open-label Phase 2 clinical trial evaluating the safety, tolerability, and efficacy of BMB-101, a highly selective 5-HT2C receptor agonist, in adult patients with classic Absence Epilepsy and Developmental Epileptic Encephalopathy (DEE).
Financial Year Ended September 30, 2025
On October 3, 2024, the Company announced the grant of 70,000 Options to employees and members of the Board of Directors. The Options are exercisable at an exercise price of $1.65 for a period of five (5) years from the date of grant.
On October 16, 2024, the Company announced positive data from the preclinical testing of BMB-201 completed with National Institute of Health pain screening (PSPP) program.
On October 21, 2024, the Company announced a collaboration with Firefly Neuroscience, Inc., an artificial intelligence company developing innovative solutions that improve brain health outcomes for patients with neurological and mental disorders, to provide a full analysis of the electroencephalogram (EEG) data in the Company’s BREAKTHROUGH study, an open-label Phase 2 clinical trial evaluating the safety, tolerability, and efficacy of BMB-101, a highly selective 5-HT2C receptor agonist, in adult patients with classic Absence Epilepsy and Developmental Epileptic Encephalopathy (DEE).
On November 4, 2024, the Company closed a non-brokered private placement of 1,612,902 Common Shares at a price of USD$21.70 per Common Share for aggregate gross proceeds of USD$35,000,000 (the “November 2024 Private Placement”).
On January 7, 2025, the Company appointed Stephen D. Collins, M.D., Ph.D., as Chief Medical Officer. Mark A. Smith, M.D., Ph.D., the Company’s former Chief Medical Officer, retired from his role but continued to serve the Company in an advisory capacity.
On February 5, 2025, the Company filed a registration statement on Form F-3 to register the 1,612,902 Common Shares issued pursuant to the November 2024 Private Placement.
On May 13, 2025, the Company announced positive findings from its DBA/2 mouse model study. BMB-101, the Company’s novel scaffold 5-HT2C Gq-protein biased agonist, demonstrated a complete elimination of drop attacks in the DBA/2 mouse model.
On August 25, 2025, the Company filed a registration statement on Form F-3 with the SEC containing a base shelf prospectus and an at-the-market offering prospectus supplement (the “Base Shelf Registration Statement”). As a result of this filing, the Company is permitted to publicly offer up to USD$250,000,000 of common shares, common share purchase warrants, or units consisting of common shares and warrants to investors in the United States during the period the Base Shelf Registration Statement is effective. On the same date, the Company entered into an equity distribution agreement (the “Equity Distribution Agreement”) providing for an at-the-market equity offering program (“ATM”) with Piper Sandler & Co. and Cantor Fitzgerald & Co. (collectively, the “Agents”). The ATM allow Bright Minds, through the Agents, to offer and sell from time to time in the United States, through the facilities of NASDAQ, or any other method permitted by law as defined under Rule 415 of the U.S. Securities Act, such number of Common Shares as would have an aggregate offering price of up to USD$100 million. The sale of Common Shares through the ATM will be made pursuant to the Base Shelf Registration Statement, which was made effective on September 2, 2025. The ATM will expire on September 2, 2028, unless terminated earlier in accordance with the terms of the Equity Distribution Agreement. The timing and extent of the use of the ATM will be at the discretion of the Company.
Current Financial Year to the date of this AIF
During the current financial year, the Company expects to continue clinical development of BMB-101 in epilepsy indications (Phase 2/3 studies), and may utilize its existing Base Shelf Registration Statement to conduct capital raises, subject to market conditions and operational needs.
On November 6, 2025, the Company announced the initiation of Prader-Willi Syndrome (“PWS”) program and exploratory Phase 2 clinical study with BMB-101 in PWS. The Company expects to advance its preclinical candidate BMB-105 to Phase 1 clinical studies.
DESCRIPTION OF THE BUSINESS
Company Overview
The Company is a biotechnology company developing innovative treatments for patients with neurological and psychiatric disorders. The Bright Minds pipeline includes novel compounds targeting key receptors in the brain to address conditions with high unmet medical need, including epilepsy, depression, and other central nervous system (CNS) disorders. The Company is focused on delivering breakthrough therapies that can transform patients’ lives. The Company has developed a unique platform of highly selective serotonergic agonists exhibiting selectivity at different serotonergic receptors. This has provided a rich portfolio of new chemical entity (NCE) programs within neurology and psychiatry.
Principal Products and Services
Serotonin (5-HT) is the most prominent neurotransmitter in the brain and modulates many biological functions. Dysfunction of serotonin receptors, transporters, and associated neurocircuits is fundamental to many diseases including epilepsies and neuro-psychiatric disorders such as depression. The class of medications known as selective serotonin reuptake inhibitors, such as Prozac®, Zoloft®, and Lexapro®, are widely used in the treatment of depression with a market of USD$14.3 Billion1. Similarly, other serotoninergic drugs are widely used in the treatment of pain (Triptans in migraine)2, Alzheimer’s and Parkinson’s disease related psychosis (Pimavanserin)3, and seizures (Fintepla)4. The off-label use of psilocybin extracts in depression and cluster headache, as well as encouraging clinical trial data with psilocybin and MDMA in depression and PTSD illustrate the potential for advancing serotoninergic therapies in neuropsychiatry and pain. The full potential of serotonin-based therapeutics has not been achieved due to the lack of medications that are selective and specific to certain serotonin receptor subtypes that are fundamental to disease pathology, without non-specific effects, or other off-target effects on other serotonin receptors in the body that are associated with cardiac toxicities and have resulted in previous drugs being withdrawn from the market.
|
1 |
Research and Markets, “Global Antidepressants Market (2020 to 2030) - COVID-19 Implications and Growth” (21 April 2020), online: Intrado GlobeNewswire <https://www.globenewswire.com/news-release/2020/04/21/2019282/0/en/Global-Antidepressants-Market-2020-to-2030-COVID-19-Implications-and-Growth.html>. |
|
2 |
Samar Nicolas & Diala Nicolas, “Triptans” (26 May 2020), online: National Center for Biotechnology Information <https://www.ncbi.nlm.nih.gov/books/NBK554507/>. |
|
3 |
Cerner Multum, “Pimavanserin” (5 February 2020), online: Drugs.com <https://www.drugs.com/mtm/pimavanserin.html>. |
|
4 |
Fintepla FDA Approval History” (accessed 5 May 2021), online: Drugs.com <https://www.drugs.com/history/fintepla.html>. |
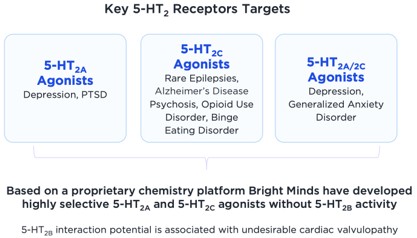
Bright Minds has a portfolio of patented, selective serotonin (5-HT2C and 5-HT2C/A-receptor subtypes) agonists that were identified using high-throughput screening methods in combination with advanced molecular modeling techniques to interrogate the interaction between the drug and its targeted receptors to increase downstream signaling while avoiding off-target effects.

The Company’s lead program is 5-HT2C selective agonist BMB-101. It is a novel scaffold 5-HT2C agonist developed using structure-based drug design. Biased agonism at the 5-HT2C receptor is one of its key features and adds another layer of functional selectivity within a well-validated target. BMB-101 works exclusively via the Gq-protein signaling pathway and avoids beta-arrestin activation, which is crucial to minimize the risk of receptor desensitization and tolerance development. This provides a novel mechanism, anti-epileptic drug designed to provide sustained seizure relief in hard-to-treat patient populations. In preclinical studies, BMB-101 has demonstrated efficacy in animal models of Dravet Syndrome and animal models of generalized seizures.
In Phase 1 clinical studies, BMB-101was tested in healthy volunteers in a Single Ascending Dose (SAD), Multiple Ascending Dose (MAD) and food-effects study. BMB-101 was demonstrated to be safe and well tolerated at all doses. No Serious Adverse Events (SAEs) were observed, and Adverse Events (AEs) were mild in nature and in line with on-target effects for serotonergic drugs.
An extensive target-engagement study was conducted using both fluid biomarkers (transient prolactin release) and physical biomarkers (Quantitative Electroencephalogram, qEEG). Both methods confirmed robust central target engagement. A qEEG signature typicalfor anti-epileptic drugs was observed, with a selective depression of EEG power at frequencies observed during epileptic seizures. Furthermore, a potentiation of frontal gamma-power was observed in this study which could indicate the potential for improved cognition.
Phase 2 clinical trials of BMB-101 were initiated in 2024 in a group of drug-resistant epilepsies including Developmental and Epileptic Encephalopathies and Absence epilepsies.
The Company plans to explore the utility of its highly selective 5-HT2C agonists in PWS, and exploratory Phase 2 study of BMB-101 in PWS patients is planned in 2026.
Lead candidates in other programs include BMB-105, novel 5-HT2C agonist, and BMB-201 and BMB-202, selective 5-HT2A agonists for neuropsychiatry and neurology indications, undergoing IND-enabling studies.
Stage of Development of Principal Products
|
Drug candidate |
Program |
Medical indications |
Status
|
|
BMB-101 |
5-HT2C agonist |
DEE
Absence Seizures
Prader Willi Syndrome |
Preclinical characterisation: IND-enabling package completed
API and Drug Product manufacturing: Completed. Optimization and drug product resupply is ongoing.
Clinical:
Phase 1 clinical trials (SAD/MAD/Food effects) completed
Phase 2 clinical trials in DEE and absence seizures patients – ongoing
Future Phase 2/3 clinical trials in epilepsy indications – preparatory activities ongoing
Phase 2 clinical trial in PWS patients – preparatory activities ongoing
|
|
BMB-105 |
5-HT2C agonist |
Prader Willi Syndrome |
Preclinical characterisation: IND-enabling package ongoing
API and Drug Product manufacturing: Ongoing
|
|
BMB-201 |
5-HT2A/2C agonist |
Pain / psychiatry indication |
Preclinical characterisation: IND-enabling package ongoing
|
|
BMB-202 |
5-HT2A agonist |
Psychiatry indication |
Preclinical characterisation: IND-enabling package ongoing
|
The Company has completed a Phase 1 clinical trial on its leading product candidate, 5-HT2C agonist, as follows:
|
Product |
Indications |
Clinical Trial |
Major Objective |
Outcome |
||
|
BMB-101 |
Undisclosed Seizure Disorder |
Phase 1 SAD, MAD and Food Effects |
● |
Safety, PK/PD and Exploratory Effect markers |
●
|
Based on safety results, BMB-101 was safe and well-tolerated by healthy subjects. |
| ● | Prolactin and qEEG changes were indicative of central target engagement of central 5-HT2c receptors by BMB-101. | |||||
|
Absence Epilepsy and DEE |
BREAKTHROUGH Study: A Phase 2 Trial of BMB-101 in Absence Epilepsy and Developmental Epileptic Encephalopathy |
● |
Assess the safety, tolerability and efficacy of BMB-101 |
● |
Ongoing study NCT06401538 | |
Production and Services
Currently, the Company has no commercial production and is in the process of developing its principal products and completing clinical trials. Upon the successful development of one of the Company’s principal products the Company will establish a method of production.
Specialized Skill and Knowledge
The nature of the Company’s business requires specialized skill and knowledge, including expertise in medicine and healthcare. The Company’s management and Scientific Advisory Board has expertise drug development and therapeutics. The required skills and knowledge to succeed in our industry are available to Bright Minds through certain members of the company’s management, directors, officers, and advisory teams. See “DIRECTORS AND OFFICERS”.
Competitive Conditions
The biotechnology and biopharmaceutical industries, and the neurological subsector, are characterized by rapid evolution of technologies, fierce competition, and strong defense of intellectual property. Any product candidates that the Company successfully develop and commercialize will have to compete with existing therapies and new therapies that may become available in the future. While the Company believes that their rational approach to drug design, along with their scientific expertise in the field of serotonergic drugs and central nervous system (CNS) function, provide the Company with competitive advantages, a wide variety of institutions, including large biopharmaceutical companies, specialty biotechnology companies, academic research departments, and public and private research institutions, are actively developing potentially competitive products and technologies. The Company’s competitors generally fall within the following categories:
|
● |
Developmental and Epileptic Encephalopathy/Epilepsy. UCB, Jazz Pharmaceuticals, H. Lundbeck A/S, Biocodex, Xenon Pharmaceuticals, Praxis Therapeutics, SK Life Science, Ovid Therapeutics, Supernus Pharmaceuticals, Harmony Biosciences, Takeda Pharmaceuticals, Eisai. |
|
● |
Prader-Willi Syndrome: Soleno Therapeutics, Aardvark Therapeutics, Rhythm Pharmaceuticals, Acadia Pharmaceuticals, Harmony Biosciences, Neuren Pharmaceuticals |
|
● |
Antidepressants and anxiolytics. AbbVie Inc., AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, GlaxoSmithKline, H. Lundbeck A/S, Johnson & Johnson, Merck, Novartis, Otsuka Pharmaceutical, Pfizer Inc., Sanofi, Takeda Pharmaceutical Company Ltd. |
Intangible Properties
Patents and Patent Applications
Kozikowski-Roth Patents
The Company has exclusively licensed a family of patents based on PCT/US2011/023535, which is co-owned by the Board of Trustees of UIC and the University of North Carolina at Chapel Hill. This family of licensed patents includes patents granted in Australia (AU Pat No 2011212930), Canada (CA Pat No 2788416), Europe (EU Pat No 2531485), Japan (JP Pat No 5810099), United States (US Pat No 8492591 and US Pat No 8754132). In addition, the Company has exclusively licensed a family of patents based on PCT/US2016/015019, which is solely owned by the Board of Trustees of UIC. This family of licensed patents includes patents applied for or granted in China (CN Publication No 107810175), Europe (EU Publication No 3250549), Hong Kong SAR (HK Publication No 1251831), and the United States (US Pat No 10407381). The latest patent to issue is US Pat No 10407381 which will expire on January 27, 2036.
These patents were based on the past research completed by Dr. Alan Kozikowski and Dr. Bryan Roth that is documented in United States publication number US20090203750A1 “5-HT2C Receptor Agonists as Anorectic Agents”. The invention related to the discovery of novel selective 5-HT2C and 5-HT2C/A agonists that could be used for the treatment of multiple neurological conditions.
On May 26, 2020, the Company entered into an option agreement (the “Roth Kozikowski Agreement”) with the Board of Trustees of UIC in which UIC granted the Company, in consideration for an option fee, an exclusive option to: (i) evaluate the inventions described in PCT/US2011/023535 and all counterpart patents related thereto and described in PCT/US2016/015019 and all counterpart patents related thereto (collectively, the “Inventions”); and (ii) obtain an exclusive license to the Inventions. On April 23, 2021, the Company and UIC entered into a First Amendment to the Roth Kozikowski Agreement for the purpose of amending certain terms in the Roth Kozikowski Agreement.
On April 23, 2021, the Company entered into an exclusive license agreement (the “Exclusive License Agreement”) with UIC pursuant to the exercise of its option under the Roth Kozikowski Agreement and the First Amendment to the Roth Kozikowski Agreement. Pursuant to the terms and conditions of the Exclusive License Agreement, UIC granted the Company an exclusive license to the Inventions (the “License”). In consideration for the License, the Company (i) paid UIC a signing fee of USD$100,000, less USD$15,000 paid by the Company pursuant to the Roth Kozikowski Agreement; and (ii) issued 63,000 Common Shares at a deemed price of $5.85 per Common Share to the UIC (part of which was received by UIC on behalf of the University of North Carolina at Chapel Hill). Additionally, the Company agreed to pay UIC a royalty on net sales of products derived from the Inventions and a portion of all revenue received by the Company from sublicensees.
The Company may terminate the Exclusive License Agreement at any time on written notice to UIC at least ninety (90) days prior to the termination date specified in the notice. The notice of termination must also include the Company’s reason for such termination. UIC may terminate the Exclusive License Agreement if the Company: (a) fails to pay any amount, or provide any other consideration, or make any report when required, and the Company does not cure such failure within ninety (90) days after receiving notice thereof; (b) is in breach of any provision of the Exclusive License Agreement not covered by (a) and the Company does not cure such failure within forty-five (45) days after receiving notice thereof; (c) is in breach of any obligations that the Company has to UIC under any other agreement between the Company and UIC and the Company does not cure such failure within ninety (90) days after receiving notice thereof, however, should the Company be aware it is unable to remedy such breach within ninety (90) days, the Company shall have the option to provide written notice to UIC after which the Company and UIC shall negotiate in good faith to determine an appropriate extension to said ninety (90) day time frame; (d) makes any materially false report and receives written notice from UIC; (e) to the extent not prohibited by applicable law commences a voluntary case as a debtor under the Bankruptcy Code, or if an involuntary case is commenced against the Company under the Bankruptcy Code, or if an order for relief shall be entered in such case, or if the same or any similar circumstance shall occur under the laws of any foreign jurisdiction; and/or (f) takes any action that purports to cause or causes any of the patent rights or technical information subject to the Exclusive License Agreement to be subject to any lien or encumbrance, and such termination shall be upon written notice to the Company.
Filed Patent Applications
Based upon molecular modeling studies in concert with data available from published research articles, the Bright Minds chemistry team designed novel analogs of psilocin that they believed would retain 5-HT2A activity while having no propensity to activate the 5‑HT2B receptors. These new chemical entities were thus anticipated to retain the brain re-booting activity of psilocin while showing no propensity to cause valvulopathy issues.
On March 12, 2020, Bright Minds filed a United States provisional application that was assigned a serial number of US 62/988,926. This patent application focused on psilocin analogs that have been decorated with functionality appropriate to achieving the goals of maintaining the desired 5-HT2A activity while being devoid of 5-HT2B activity. On March 12, 2021, Bright Minds filed a Patent Cooperation Treaty patent application that claims priority to US 62/988,926. Such Patent Cooperation Treaty patent application was assigned a serial number of PCT/CA2021/050336. In September 2022, PCT/CA2021/050336 entered the national phase in the United States of America; the United States application, which has been assigned a serial number of 17/911,022, is currently pending. In October 2022, PCT/CA2021/050336 entered the national phase in the European Union; the European Union application, which has been assigned a serial number of 21768153.5, is currently pending.
On May 26, 2021, Bright Minds filed a United States provisional application that was assigned a serial number of US 63/193,062. This patent application focused on substitutions at a particular position on an indole structure. On May 25, 2022, Bright Minds filed a Patent Cooperation Treaty patent application that claims priority to US 63/193,062. Such Patent Cooperation Treaty patent application has been assigned a serial number of PCT/CA2022/050833. PCT/CA2022/050833 has entered the national/regional phase in the United States of America (assigned application number 18/562,587), Canada (assigned application number 3,219,940), Japan (assigned application number 2023-573064), the European Union (assigned application number 22810004.6), South Korea (assigned application number 10-2023-7044608), and China (assigned application number 202280052820.8).
On January 4, 2022, Bright Minds filed a United States provisional application that has been assigned a serial number of US 63/296,430. This patent application focuses on phenethylamine compounds. On November 4, 2022, Bright Minds filed a second United States provisional application focused on phenethylamine compounds; this provisional application has been assigned a serial number of US 63/422,730. On January 4, 2023, Bright Minds filed a Patent Cooperation Treaty patent application that claims priority to US 63/296,430 and US 63/422,730. Such Patent Cooperation Treaty patent application has been assigned a serial number of PCT/CA2023/050003. PCT/CA2022/050003 has entered the national/regional phase in Canada (assigned application number 3,242,928), the United States of America (assigned application number 18/726,389), Australia (assigned application number 2023205941), Japan (assigned application number 2024-540591), the European Union (assigned application number 23736951.7), South Korea (assigned application number 10-2024-7026278), and China (assigned application number 202380022140.6).
On May 6, 2022, Bright Minds filed a United States provisional application that has been assigned a serial number of US 63/338,889. This patent application focuses on substitutions at a particular position on an indole structure. On May 2, 2023, Bright Minds filed a Patent Cooperation Treaty patent application that claims priority to US 63/338,889. Such Patent Cooperation Treaty patent application has been assigned a serial number of PCT/CA2023/050595. PCT/CA2023/050595 has entered the national/regional phase in Canada (assigned application number 3,252,369), Japan (application number to be assigned), Australia (assigned application number 2023264112), the United States of America (assigned application number 18/863,516), and the European Union (assigned application number 23799069.2). Bright Minds also intends for PCT/CA2023/050595 to enter the national phase in China, and the deadline to enter the Chinese national phase is January 6, 2025.
Bright Minds is currently listed as an applicant in the following publicly disclosed matters:
|
Patent Application Number |
Region |
Title |
Inventors |
Applicant |
Filing Date |
Status |
|
17/911,0225 |
USA |
3-(2-(AMINOETHYL)-INDOL-4-OL DERIVATIVES, METHODS OF PREPARATION THEREOF, AND THE USE AS 5-HT2 RECEPTOR MODULATORS |
Alan KOZIKOWSKI Gideon SHAPIRO Werner TUECKMANTEL John McCORVY |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
March 12, 2021 (national phase entry of PCT/CA2021/050336 having an international filing date of March 12, 2021, which claims priority to US 62/988,926 filed March 12, 2020) |
Pending |
|
21768153.56 |
European Union |
3-(2-(AMINOETHYL)-INDOL-4-OL DERIVATIVES, METHODS OF PREPARATION THEREOF, AND THE USE AS 5-HT2 RECEPTOR MODULATORS |
Alan KOZIKOWSKI Gideon SHAPIRO Werner TUECKMANTEL John McCORVY |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
March 12, 2021 (national phase entry of PCT/CA2021/050336 having an international filing date of March 12, 2021, which claims priority to US 62/988,926 filed March 12, 2020) |
Pending |
|
5 |
Pursuant to an inter-institutional agreement effective as of December 16, 2022, entered into between the Company and The Medical College of Wisconsin, Inc. (“MCW”), MCW is responsible for prosecuting and maintaining this application and negotiating licenses relating to the application. |
| 6 |
See note 2. |
|
18/562,587 |
USA |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
3,219,940 |
Canada |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
2023-573064 |
Japan |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
22810004.6 |
European Union |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
10-2023-7044608 |
South Korea |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
202280052820.8 |
China |
Heterocyclic Compounds and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL John MCCORVY Uros LABAN |
Bright Minds Biosciences Inc.; The Medical College of Wisconsin, Inc. |
May 25, 2022 (national phase entry of PCT/CA2022/050833 having an international filing date of May 25 2022, which claims priority to US 63/193,062 filed May 26, 2021 and US 63/309,735 filed February 14, 2022) |
Pending |
|
3,242,928 |
Canada |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
18/726,389 |
USA |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
July 2, 2024 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
2023205941 |
Australia |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
2024-540591 |
Japan |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
23736951.7 |
European Union |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
10-2024-7026278 |
South Korea |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
202380022140.6 |
China |
Phenethylamines and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
January 4, 2023 (national phase entry of PCT/CA2023/050003 having an international filing date of January 4, 2023, which claims priority to US63/296,430 filed January 4, 2022 and US63/422,730 filed November 4, 2022) |
Pending |
|
3,252,369 |
Canada |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
|
2024-565127 |
Japan |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
|
2023264112 |
Australia |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
|
18/863,516 |
USA |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
|
23799069.2 |
European Union |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
|
202380051305.2 |
China |
Azepinoindoles and Methods of Preparation Thereof |
Alan KOZIKOWSKI Werner TUECKMANTEL |
Bright Minds Biosciences Inc. |
May 2, 2023 (national phase entry of PCT/CA2023/050595 having an international filing date of May 2, 2023, which claims priority to US 63/338,889 filed May 6, 2022) |
Pending |
The Company continues research and development (“R&D”) related to highly selective 5-HT2 agonists for neurological and neuropsychiatric indications. As a part of its ongoing R&D programs, the Company has filed and anticipates that it will continue to file additional patents to protect its intellectual property.
Trademarks
Bright Minds has the following trademark registrations:
|
Trademark |
Country |
Application Number (Registration Number) |
Filing Date (Registration Date) |
Status |
|
BRIGHT MINDS |
Canada |
2,016,213 (TMA1202388) |
2020-03-06 (2023-10-11) |
Registered |
|
BRIGHT MINDS |
United States of America |
90/245,748 (7,424,235) |
2020-10-09 (2024-06-25) |
Registered |
Web Domains
Bright Minds has use and control over the following domain name: brightmindsbio.com.
Government Regulation
Regulatory Framework
Drug products must be approved by the appropriate governing body before it can be sold in that country or area. The FDA approves products for the United States market and Health Canada approves products for the Canadian market. The European Medicines Agency approves products for the European Union. While the process by which products are approved by the FDA and Health Canada is very similar, each regulatory body has its own unique requirements for a product. In both cases, the development of a product through to approval can be a lengthy process and, in some cases, can take over 10 years. While early studies conducted in one jurisdiction will usually be accepted in the other, further and somewhat modified studies may be required to have a product approved in another jurisdiction.
Canadian Government Regulation
Drug products in Canada are regulated by Health Canada under the Food and Drugs Act and the Food and Drugs Regulations. Health Canada regulates, among other things, research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, advertising and promotion of any product candidates or commercial products. For a drug product to be approved in Canada, it must provide sufficient evidence of safety, efficacy and chemical quality based on preclinical investigation and Phase I, II and III clinical trials using approved and compliant manufacturing and clinical sites.
To obtain approval to market a drug in Canada, a sponsor usually requests a pre-submission meeting with the review division of Health Canada responsible for the therapeutic field. If the meeting is granted, the sponsor must submit a Pre-Submission Information package to the Therapeutic Products Directorate (“TPD”) to meet with the review division. This process occurs prior to submitting the New Drug Submission (“NDS”) application. The purpose of the pre-submission meeting is to review the evidence (non-clinical and clinical research, quality information, indication) that will be submitted in the NDS application.
During the drug development process, the sponsor prepares study reports. Once the sponsor releases the last study required for the submission, the sponsor completes the NDS application and submits it to the TPD. Prior to submitting the NDS and, if applicable, based on the intended use of the product in the identified patient population, the sponsor may submit in advance a request for priority review status.
After submitting the NDS application, the file undergoes a screening process prior to being accepted for review. TPD has 45 calendar days from receipt to complete the screening review process. If granted a priority review, the screening period is reduced to 25 calendar days.
After a comprehensive review of an NDS application, Health Canada will issue a Notice of Compliance if the product is approved or a Notice of Non-Compliance if further questions remain. If a Notice of Compliance is issued, a Drug Identification Number is also issued that is required to be printed on each label of the product, as well as the final version of the Product Monograph that has been agreed to between Health Canada and the sponsor.
The average target time for reaching a first decision on an NDS is 300 calendar days, unless the submission has received a priority review in which case the time is 180 calendar days. Fees are levied for a review of an NDS application.
The regulatory approval process is generally lengthy and expensive, with no guarantee of a positive result. Once approved, Health Canada continues to monitor the product and license holders have obligations related to reporting to Health Canada, record keeping and ensuring continued safety and efficacy of the product. Failure to comply with any of the above applicable regulations, regulatory authorities or other requirements may result in civil or criminal penalties, recall or seizure of products, injunctive relief including partial or total suspension of production, or withdrawal of a product from the market.
United States Government Regulation
In the United States, the FDA regulates drugs under the FDCA, and its implementing regulations, and biologics under the FDCA and the Public Health Service Act, and its implementing regulations. FDA approval is required before any new unapproved drug or biologic or dosage form, including a new use of a previously approved drug, can be marketed in the United States. In some cases, changes to aspects of an approved drug product also require pre-approval prior to implementation of these changes. Drugs and biologics are also subject to other federal, state, and local statutes and regulations. If Bright Minds fails to comply with applicable FDA or other requirements at any time during the product development process, clinical testing, the approval process or after approval, Bright Minds may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, civil monetary penalties or criminal prosecution. Any FDA enforcement action could have a material adverse effect on Bright Minds.
The process required by the FDA before drug products may be marketed in the United States generally involves the following:
|
● |
Completion of extensive preclinical laboratory tests and preclinical animal studies, some performed in accordance with the GLP regulations; |
|
● |
submission to the FDA of an Investigational New Drug (“IND”) Application, which must be reviewed by the FDA and become active before human clinical trials may begin and must be updated annually; |
|
● |
approval by an independent review board (“IRB”) or ethics committee representing each clinical site before each clinical trial may be initiated; |
|
● |
performance of adequate and well-controlled human clinical trials conducted under Good Clinical Practices (“GCP”) to establish the safety and efficacy of the product candidate for each proposed indication; |
|
● |
preparation of and submission to the FDA of a New Drug Application (“NDA”) or Biologics License Application (“BLA”) after completion of all pivotal clinical trials; |
|
● |
a determination by the FDA within 60 days of its receipt of an NDA or BLA to file the application for review; |
|
● |
potential review of the product application by an FDA advisory committee, where appropriate and if applicable; |
|
● |
satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities where the proposed product is produced to assess compliance with current GMP; |
|
● |
a potential FDA audit of the preclinical research and clinical trial sites that generated the data in support of the NDA or BLA; and |
|
● |
FDA review and approval of an NDA or BLA prior to any commercial marketing or sale of the product in the United States. |
The preclinical research, clinical testing and approval process require substantial time, effort, and financial resources, and Bright Minds cannot be certain that any approvals for the Company’s product candidates will be granted on a timely basis, if at all. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans in clinical trials. The central focus of an IND submission is on the general investigational plan and the protocol(s) for human clinical trials. The IND also includes results of animal studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational new drug.
An IND must become effective before human clinical trials may begin in the US. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to the proposed clinical trials. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before clinical trials can begin. Accordingly, submission of an IND may or may not result in the FDA allowing clinical trials to commence. As drug product programs continue in development, clinical trial protocols, additional preclinical testing results, and manufacturing information is submitted with the IND to facilitate discussions with the FDA and approval of additional clinical trials.
Clinical Trials
Clinical trials involve the administration of the IND to human subjects under the supervision of qualified investigators in accordance with GCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the efficacy criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. Additionally, approval must also be obtained from each clinical trial site’s IRB or ethics committee, before the trials may be initiated, and the IRB or ethics committee must monitor the trial until completed. All subjects must provide informed consent prior to participating in the trial. There are also requirements governing the reporting of ongoing clinical trials and clinical trial results to public registries.
The clinical investigation of a drug is generally divided into three or four phases. Although the phases are usually conducted sequentially, they may overlap or be combined.
|
● |
Phase I. The drug is initially introduced into healthy human subjects or, in some cases, patients with the target disease or condition. These studies are designed to evaluate the safety, tolerance, metabolism, pharmacokinetic and pharmacologic actions of the investigational new drug in humans, and the side effects associated with increasing doses. |
|
● |
Phase II. The drug is administered to a limited patient population to evaluate safety and optimal dose levels for safety and efficacy, identify possible adverse side effects and safety risks, and preliminarily evaluate efficacy. |
|
● |
Phase III. The drug is administered to an expanded patient population, generally at geographically dispersed clinical trial sites to generate sufficient data to statistically evaluate dose levels, clinical effectiveness and safety, to establish the overall benefit-risk relationship of the IND product, and to provide an adequate basis for physician labeling. |
|
● |
Phase IV. In some cases, the FDA may conditionally approve an NDA or BLA for a drug product with the sponsor's agreement to conduct additional clinical trials after approval. In other cases, a sponsor may voluntarily conduct additional clinical trials after approval to gain more information about the drug. Such post-approval studies are typically referred to as Phase IV clinical trials. |
Clinical trial sponsors must also report to the FDA, within certain timeframes: (i) serious and unexpected adverse reactions, (ii) any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator’s brochure, or (iii) any findings from other studies or animal testing that suggest a significant risk in humans exposed to the product candidate. The FDA, the IRB, the ethics committee or the clinical trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether or not a trial may move forward at designated check points based on access to certain data from the trial.
The clinical trial process can take years to complete, and there can be no assurance that the data collected will support FDA approval or licensing of the product. Results from one trial are not necessarily predictive of results from later trials. The Company may also suspend or terminate a clinical trial based on evolving business objectives and/or competitive climate.
Submission of an NDA or BLA to the FDA
Assuming successful completion of all required preclinical studies and clinical testing in accordance with all applicable regulatory requirements, detailed IND product information is submitted to the FDA in the form of an NDA or BLA requesting approval to market the product for one or more indications. Under federal law, the submission of most NDAs and BLAs is subject to an application user fee. Applications for Oppositional Defiant Disorder products are exempted from the NDA and BLA application user fee, unless the application includes an indication for other than a rare disease or condition and may be exempted from product and establishment user fees under certain conditions. An NDA or BLA must include all relevant data available from pertinent preclinical studies and clinical trials, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data comes from company-sponsored clinical trials intended to test the safety and effectiveness of a use of a product, and may also come from several alternative sources, including clinical trials initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and effectiveness of the investigational new drug product to the satisfaction of the FDA.
Once an NDA or BLA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by the FDA’s requests for additional information or clarification. Before approving an NDA or BLA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are following current GMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA or BLA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP and related regulations.
The FDA is required to refer an NDA or BLA for a novel drug (in which no active ingredient has been approved in any other application) to an advisory committee or explain why such referral was not made. Typically, an advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and the conditions thereof. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
The FDA’s Decision on an NDA or BLA
After the FDA evaluates the NDA or BLA and conducts inspections of manufacturing facilities where the product will be produced, the FDA will issue either an approval letter or a complete response letter (“Complete Response Letter”). An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete, and the application is not ready for approval. To satisfy deficiencies identified in a Complete Response Letter, additional clinical data and/or an additional Phase III clinical trial(s), and/or other significant, expensive and time-consuming requirements related to clinical trials, preclinical studies or manufacturing may be required for the drug product.
Even if such additional information is submitted, the FDA may ultimately decide that the NDA or BLA does not satisfy the criteria for approval. The FDA could also approve the NDA or BLA with a risk evaluation and mitigation strategy, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA may also conditionally approve a drug product subject to, among other things, changes to proposed labeling, development of adequate controls and specifications, or a commitment to conduct one or more post-market studies or clinical trials. Such post-market testing may include Phase IV clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization. New government requirements, including those resulting from new legislation, may be established during the review process, or the FDA’s policies may change, which could delay or prevent regulatory approval of the Company’s products under development.
The Company has numerous options as it relates to contract manufactures of GMP (good manufacturing products) grade active pharmaceutical ingredients and finished products. The Company does not expect to encounter any issues sourcing raw materials nor do we foresee material volatility in raw materials and finished good pricing.
Cycles
The Company’s business is not cyclical or seasonal.
Economic Dependence
The Company is not substantially dependent on any contracts.
Changes to Contracts
The Company does not expect any aspect of the Company’s business to be affected by renegotiation or termination of contracts.
Environmental Protection
The Company’s business does not materially impact environmental conditions. The Company does not expect that there will be any financial or operational effects as a result of environmental protection requirements on its capital expenditures, profit or loss, or its competitive position in the current fiscal year or in future years.
Employees
As at September 30, 2025, the Company’s wholly-owned subsidiary, Bright Minds Biosciences LLC, had one employee and the Company as a whole had less than 25 full time equivalent staff.
Foreign Operations
The Company conducts certain clinical development activities outside of Canada and the United States, specifically in Australia where, as part of its clinical strategy the Company has sponsored and executed clinical drug trials. These are completed through arrangements with local contract research organizations (CROs), clinical sites, and service providers. Activities include site initiation, patient screening and enrollment, trial administration, clinical monitoring, data management, and handling of clinical trial material in accordance with Australian regulatory requirements.
These foreign activities are material to the Company’s business, and the Company is dependent on the continued availability and performance of Australian CROs, clinical sites, and investigators, as well as the timely receipt of ethics and regulatory approvals. The Company is subject to risks inherent in foreign operations, including differences in regulatory requirements and review timelines, changes in local laws or regulatory interpretations, availability of eligible patient populations, currency fluctuation and inflation, logistical challenges related to the sourcing, shipment and storage of the clinical trial materials, potential disruptions arising from public health events, geopolitical developments, or natural disasters; and counterparty performance, including CRO or site non-compliance with protocol. The Company mitigates these risks through a combination of contractual and operational controls. These include engaging reputable CROs and trial sites and operating in well established jurisdictions such as Australia.
At the date of this AIF, the Company believes that its Australian clinical activities are appropriately resourced and operating in accordance with applicable regulatory and ethical requirements. Nonetheless, there can be no assurance that the risks inherent in foreign operations will not materialize or that any resulting delays or additional costs will not be material. Any changes in local regulations or shifts in political conditions may adversely affect the Company’s ability to conduct clinical drug trials. The Company will continue to evaluate its reliance on Australian operations in light of development needs, regulatory feedback, and operational performance, and may expand or contract the scope of its foreign activities accordingly.
Lending
The Company has no lending operations.
Bankruptcy and Similar Procedures
There have been no bankruptcy or receivership proceedings against the Company.
Reorganizations
The Company has not completed any reorganizations of it or its subsidiaries within the three most recently completed financial years, and does not anticipate completing any such reorganizations in the current financial year.
Social or Environmental Policies
The Board has adopted a Code of Business Conduct and Ethics (the “Code of Ethics”) that applies to all of our employees and officers, including our principal executive officer, principal financial officer, principal accounting officer or controller or persons performing similar functions. The Board is responsible for monitoring compliance with the Code of Ethics.
RISK FACTORS
The following are certain risk factors relating to the business and securities of the Company. The following information is a summary only of certain risk factors and is qualified in its entirety by reference to, and must be read in conjunction with, the detailed information appearing elsewhere in this AIF. These risks and uncertainties are not the only ones facing the Company. Additional risks and uncertainties not presently known to the Company, or that the Company currently deems immaterial, may also impair the operations of the Company. If any such risks actually occur, the business, financial condition and/or liquidity and results of operations of the Company could be materially adversely affected.
Risks Related to the Business of the Company
The Company has a limited operating history and have not yet generated any revenues
The Company has a limited history of operations which makes evaluating the Company’s business and future prospects difficult. As such, the Company is subject to many risks common to such enterprises, including under-capitalization, cash shortages, limitations with respect to personnel, financial and other resources and lack of revenues. There is no assurance that the Company will be successful in achieving a return on shareholders’ investment and the likelihood of the Company’s success must be considered in light of the Company’s early stage of operations.
The Company’s actual financial position and results of operations may differ materially from the expectations of the Company’s management
The Company’s actual financial position and results of operations may differ materially from the Company’s management’s expectations. The Company has experienced some changes in its operating plans and certain delays in its plans. As a result, the Company’s revenue, net income and cash flow may differ materially from the Company’s management’s expected revenue, net income and cash flow. The process for estimating the Company’s revenue, net income and cash flow requires the use of judgment in determining the appropriate assumptions and estimates. These estimates and assumptions may be revised as additional information becomes available and as additional analyses are performed. In addition, the assumptions used in planning may not prove to be accurate, and other factors may materially affect the Company’s financial condition or results of operations.
The Company may not be successful in its efforts to identify, license or discover additional product candidates
Although a substantial amount of the Company’s effort will focus on the continued research, pre-clinical testing, clinical trials, and potential approval and commercialization of its existing product candidates, the success of its business also depends in part upon its ability to identify, license or discover additional product candidates. The Company’s research programs or licensing efforts may fail to yield additional product candidates for clinical development for a number of reasons, including but not limited to the following:
|
● |
the Company’s research or business development methodology or search criteria and process may be unsuccessful in identifying potential product candidates; |
|
● |
the Company may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; |
|
● |
the Company’s product candidates may not succeed in pre-clinical or clinical testing; |
|
● |
the Company’s product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval; |
|
● |
competitors may develop alternatives that render the Company’s product candidates obsolete or less attractive; |
|
● |
product candidates the Company develops may be covered by third parties’ patents or other exclusive rights; |
|
● |
the market for a product candidate may change during the Company’s program so that such a product may become unreasonable to continue to develop; |
|
● |
a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
|
● |
a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors. |
If any of these events occurs, the Company may be forced to abandon its development efforts to identify, license or discover additional product candidates, which would have a material adverse effect on its business and could potentially cause the Company to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. The Company may focus its efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
The Company may be required and have not yet obtained regulatory approvals, licenses, and permits in the jurisdictions where its products or technologies are being researched, developed or commercialized, which failure to obtain such regulatory approvals, licenses and permits will likely have a material adverse effect on the Company’s business, financial condition and results of operations
The Company, or its service providers, may be required to obtain and maintain certain permits, licenses, and approvals in the jurisdictions where the Company’s products or technologies are being researched, developed, and/or commercialized. The Company has not obtained regulatory approval for any product candidate, and it is possible that none of the Company’s existing product candidates or any future product candidates will ever obtain regulatory approval. There can be no assurance that the Company will be able to obtain or maintain any necessary licenses, permits, or approvals. Any material delay or inability to receive these items is likely to delay and/or inhibit the Company’s ability to conduct our business, and would have an adverse effect on its business, financial condition, and results of operations. In particular, the Company will require approval from the FDA and equivalent organizations in other countries before any of the Company’s products can be marketed. There is no assurance that such approvals will be forthcoming. Furthermore, the exact nature of the studies these regulatory agencies will require is not known and can be changed at any time by the regulatory agencies, increasing the financing risk and potentially increasing the time to market we face, which could adversely affect the Company’s business, financial condition or results of operations.
The Company may encounter substantial delays or difficulties with its clinical trials, which could have a material adverse effect on the Company’s financial condition and results of operations
The Company may not commercialize, market, promote or sell any product candidate without first obtaining marketing approval from the FDA or comparable foreign regulatory authorities, and the Company may never receive such approvals. It is impossible to predict when or if any of the Company’s product candidates will prove effective or safe in humans and if or when the Company will receive regulatory approval. Before obtaining marketing approval from regulatory authorities for the sale of its product candidates, the Company must complete pre-clinical development and then conduct extensive clinical trials to demonstrate the safety and efficacy of its product candidates. Clinical testing is expensive, difficult to design and implement, can take many years to complete and is uncertain as to outcome. A failure of one or more clinical trials can occur at any stage of testing. Moreover, pre-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that have believed their product candidates performed satisfactorily in pre-clinical studies and clinical trials have nonetheless failed to obtain marketing approval of their products.
The Company may experience numerous unforeseen events prior to, during, or as a result of, clinical trials that could delay or prevent the Company’s ability to receive marketing approval or commercialize current and any future product candidates, including:
|
● |
delays in reaching a consensus with regulatory authorities on design or implementation of clinical trials; |
|
● |
regulators or institutional review boards, or IRBs, may not authorize us or our investigators to commence a clinical trial or conduct a clinical trial at a prospective trial site; |
|
● |
delays in reaching agreement on acceptable terms with prospective clinical research organizations and clinical trial sites; |
|
● |
clinical trials of the Company’s product candidates may produce negative or inconclusive results; |
|
● |
imposition of a clinical hold by regulatory authorities as a result of a serious adverse event, concerns with a class of product candidates or after an inspection of the Company’s clinical trial operations, trial sites or manufacturing facilities; |
|
● |
occurrence of serious adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
|
● |
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; or |
|
● |
the Company may decide, or regulators may require us, to conduct additional clinical trials or abandon product development programs. |
Any inability to successfully complete pre-clinical and clinical development could result in additional costs to the Company or impair the Company’s ability to generate revenue. In addition, if the Company makes manufacturing or formulation changes to its product candidates, the Company may need to conduct additional testing to bridge its modified product candidate to earlier versions. Clinical trial delays could also shorten any periods during which the Company may have the exclusive right to commercialize its product candidates, if approved, or allow competitors to bring competing drugs to market before the Company, which could impair the Company’s ability to successfully commercialize its product candidates and may harm the Company’s business, financial condition, results of operations and prospects.
Additionally, if the results of the Company’s clinical trials are inconclusive or if there are safety concerns or serious adverse events associated with product candidates, the Company may:
|
● |
be delayed in obtaining marketing approval, if at all; |
|
● |
obtain approval for indications or patient populations that are not as broad as intended or desired; |
|
● |
obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
|
● |
be subject to additional post-marketing testing requirements; |
|
● |
be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing requirements; |
|
● |
be subject to the addition of labeling statements, such as warnings or contraindications; |
|
● |
be sued; or |
|
● |
experience damage to its reputation. |
The Company’s product development costs will also increase if the Company experience delays in testing or obtaining marketing approvals. The Company does not know whether any of the Company pre-clinical studies or clinical trials will begin as planned, need to be restructured or be completed on schedule, if at all.
Further, the Company, the FDA or an IRB may suspend the Company’s clinical trials at any time if it appears that the Company or its collaborators are failing to conduct a trial in accordance with regulatory requirements, such as the FDA’s current GCP (as defined herein), that the Company is exposing participants to unacceptable health risks, or if the FDA finds deficiencies in the Company’s INDs, or in the conduct of these trials. Therefore, the Company cannot predict with any certainty the schedule for commencement and completion of future clinical trials. If the Company experiences delays in the commencement or completion of its clinical trials, or if the Company terminates a clinical trial prior to completion, the commercial prospects of the Company’s product candidates could be negatively impacted, and the Company’s ability to generate revenues from its product candidates may be delayed.
Clinical trials are expensive, time consuming and difficult to design and implement, which could have a material adverse effect on the Company’s business, financial condition or results of operations
The Company’s product candidates will require clinical testing before the Company can submit an NDA for regulatory approval. The Company cannot predict with any certainty if or when the Company might submit an NDA for regulatory approval for any of its product candidates or whether any such NDA will be approved by the FDA. Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. For instance, the FDA may not agree with the Company’s proposed endpoints for any future clinical trial of the Company’s product candidates, which may delay the commencement of the Company’s clinical trials. The clinical trial process is also time consuming. Furthermore, failure can occur at any stage, and the Company could encounter problems that cause it to abandon or repeat clinical trials, which could have a material adverse effect on the Company’s business, financial condition or results of operations.
The Company’s current and future clinical trials or those of the Company’s current or future collaborators may reveal significant adverse events not seen in the Company’s pre-clinical and non-clinical studies and may result in a safety profile that could inhibit regulatory approval or market acceptance of any of the Company’s product candidates
Before obtaining regulatory approvals for the commercial sale of any products, the Company must demonstrate through lengthy, complex and expensive pre-clinical studies and clinical trials that its product candidates are both safe and effective for use in each target indication. Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. There is typically an extremely high rate of attrition for product candidates proceeding through clinical trials. Product candidates in later stages of clinical trials also may fail to show the desired safety and efficacy profile despite having progressed through non-clinical studies and initial clinical trials. If the results of the Company’s ongoing or future pre-clinical studies and clinical trials are inconclusive with respect to the safety and efficacy of the Company’s product candidates, if the Company does not meet the clinical endpoints with statistical and clinically meaningful significance, or if there are safety concerns associated with the Company’s product candidates, the Company may be prevented from or delayed in obtaining marketing approval for such product candidates.
In some instances, there can be significant variability in safety or efficacy results between different clinical trials of the same product candidate due to numerous factors, including changes in trial procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the clinical trial protocols, and the rate of dropout among clinical trial participants. Results of the Company’s trials could reveal a high and unacceptable severity and prevalence of side effects. Further, the Company’s product candidates could cause undesirable side effects in clinical trials related to on-target toxicity. If on-target toxicity is observed, or if the Company’s product candidates have characteristics that are unexpected, the Company may need to abandon their development or limit development to narrower uses or subpopulations in which the undesirable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective. Most product candidates that commence clinical trials are never approved as products and there can be no assurance that any of the Company’s current or future clinical trials will ultimately demonstrate positive results or support further clinical development of any of the Company’s product candidates.
If significant adverse events or other side effects are observed in any of the Company’s current or future clinical trials, the Company may have difficulty recruiting patients to its clinical trials, patients may drop out of the Company’s trials, or the Company may be required to abandon the trials or the development efforts of one or more product candidates altogether. The Company, the FDA or other applicable regulatory authorities may suspend or terminate clinical trials of a product candidate at any time for various reasons, including a belief that subjects in such trials are being exposed to unacceptable health risks or adverse side effects. Even if the side effects do not preclude the product from obtaining or maintaining marketing approval, undesirable side effects may inhibit market acceptance of the approved product due to its tolerability versus other therapies. Any of these developments could materially harm the Company’s business, financial condition, results of operations and prospects.
The Company has limited experience in completing clinical trials and has only completed one phase one drug trial to date
In July 2023, the Company completed the first Phase 1 clinical trial for its lead compound, BMB-101. Subsequently, in September 2024, the Company initiated the Phase 2 clinical trial for BMB-101. However, the Company has not yet demonstrated an ability to obtain regulatory approval, manufacture a commercial-scale product or arrange for a third party to do so on behalf of the Company, or conduct sales and marketing activities necessary for successful commercialisation of a product candidate. The Company may not be able to file an IND for BMB-101 or any of its other product candidates on the timelines the Company expects, if at all. For example, the Company may experience manufacturing delays with IND-enabling studies. Moreover, the Company cannot be sure that submission of an IND will result in the FDA allowing further clinical trials to begin, or that, once begun, issues will not arise that require the Company to suspend or terminate clinical trials. Commencing each of these clinical trials is subject to finalizing the trial design based on discussions with the FDA and other regulatory authorities. Any guidance the Company receives from regulatory authorities is subject to change. For example, a regulatory authority could change its position, including on the acceptability of the Company’s trial designs or the clinical endpoints selected, which may require the Company to complete additional clinical trials or impose stricter approval conditions than currently expected.
If the Company is required to conduct additional pre-clinical studies or clinical trials or other testing of its product candidates beyond those that are currently contemplated by the Company, if the Company is unable to successfully complete clinical trials of its product candidates or other testing, if the results of these trials or tests are not positive or are only modestly positive or if there are safety concerns, the Company may:
|
● |
be delayed in obtaining marketing approval for its product candidates; |
|
● |
not obtain marketing approval at all; |
|
● |
obtain approval for indications or patient populations that are not as broad as intended or desired; |
|
● |
be subject to post-marketing testing requirements; or |
|
● |
have the product removed from the market after obtaining marketing approval. |
If the Company experiences delays or difficulties in the enrolment of patients in clinical trials, the Company’s receipt of regulatory approvals could be delayed or prevented
The Company may not be able to initiate or continue clinical trials for its product candidates if the Company is unable to locate and enrol a sufficient number of eligible patients to participate in these trials as required by the FDA or similar regulatory authorities outside the United States. In particular, because the Company is deploying its drug discovery platform across a broad target space, the Company’s ability to enrol eligible patients may be limited or may result in slower enrolment than the Company anticipates. For example, because some of the Company’s product candidates target rare diseases, the Company may have difficulty enrolling a sufficient number of eligible patients or enrolment may be slower than anticipated. In addition, some of the Company’s competitors have ongoing clinical trials for product candidates that treat the same indications as the Company’s product candidates, and patients who would otherwise be eligible for the Company’s clinical trials may instead enrol in clinical trials of the Company’s competitors’ product candidates. The Company may not be able to identify, recruit and enrol a sufficient number of patients to complete its clinical studies for a number of reasons, including:
|
● |
the severity of the disease under investigation; |
|
● |
the eligibility criteria and overall design of the clinical trial in question; |
|
● |
the perceived risks and benefits of the product candidate under study; |
|
● |
clinicians’ and patients’ perceptions as to the potential advantages of the product candidate being studied in relation to other available therapies, including any new products that may be approved for the indications the Company is investigating; |
|
● |
the ability to obtain and maintain patient consents; |
|
● |
the efforts to facilitate timely enrolment in clinical trials; |
|
● |
the patient referral practices of physicians; |
|
● |
the size and nature of the patient population required for analysis of the trial’s primary endpoints; |
|
● |
the ability to monitor patients adequately during and after treatment; |
|
● |
the proximity and availability of clinical trial sites for prospective patients; |
|
● |
the risk that patients enrolled in clinical trials will drop out of the clinical trials before completion of their treatment; and |
|
● |
factors that the Company may not be able to control, such as potential future pandemics that may limit patients, principal investigators, staff or clinical site availability. |
Success in pre-clinical studies or clinical trials may not be predictive of results in future clinical trials
Positive results from early pre-clinical studies and clinical trials of the Company’s product candidates are not necessarily predictive of the results of later pre-clinical studies and any future clinical trials of the Company’s product candidates. Even if the Company is able to complete its planned pre-clinical studies and clinical trials of its product candidates according to the Company’s current development timeline, the results from such pre-clinical studies and clinical trials of the Company’s product candidates may not be replicated in subsequent pre-clinical studies or clinical trial results. If the Company cannot replicate such positive results in its later pre-clinical studies and future clinical trials, the Company may be unable to successfully develop, obtain regulatory approval for and commercialise its product candidates.
Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials after achieving positive results in early-stage development, and the Company cannot be certain that it will not face similar setbacks. These setbacks have been caused by, among other things, pre-clinical and other non-clinical findings made while clinical trials were underway, or safety or efficacy observations made in pre-clinical studies and clinical trials, including previously unreported adverse events. Moreover, pre-clinical, non-clinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in pre-clinical studies and clinical trials nonetheless failed to obtain FDA approval.
Additionally, future clinical trials that the Company may plan might utilise an “open-label” trial design. An “open-label” clinical trial is one where both the patient and investigator know whether the patient is receiving the investigational product candidate or either an existing approved drug or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. Open-label clinical trials may be subject to a “patient bias” where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favourably given this knowledge. The results from an open-label trial may not be predictive of future clinical trial results with any of the Company’s product candidates for which the Company includes an open-label clinical trial when studied in a controlled environment with a placebo or active control.
Interim, “topline,” and preliminary data from the Company’s clinical trials that the Company announces or publishes from time to time may change as more patient data becomes available and are subject to audit and verification procedures that could result in material changes in the final data
From time to time, the Company may publicly disclose preliminary or topline data from its clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change following a more comprehensive review of the data related to the particular study or trial. The Company may also make assumptions, estimations, calculations and conclusions as part of the Company’s analyses of data, and the Company may not have received or had the opportunity to fully and carefully evaluate all data. As a result, the topline or preliminary results that the Company reports may differ from future results of the same studies, or different conclusions or considerations may qualify such results, once additional data have been received and fully evaluated. Topline data also remains subject to audit and verification procedures that may result in the final data being materially different from the preliminary data the Company initially announces or publishes. As a result, topline data should be viewed with caution until the final data are available. From time to time, the Company may also disclose interim data from its clinical trials. Interim data from clinical trials that the Company may complete are subject to the risk that one or more of the clinical outcomes may materially change as patient enrolment continues and more patient data become available or as patients from the Company’s clinical trials continue other treatments for their disease. Adverse differences between preliminary or interim data and final data could significantly harm the Company’s business prospects.
If the interim, topline, or preliminary data that the Company reports differs from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, the Company’s ability to obtain approval for, and commercialise, its product candidates may be harmed, which could harm the Company’s business, financial condition, results of operations and prospects. In addition, the information the Company chooses to publicly disclose regarding a particular clinical trial is based on what is typically extensive information, and investors may not agree with what the Company determines is material or otherwise appropriate information to include in its disclosure.
The Company may not be successful in its efforts to identify, license or discover additional product candidates, which may have a material adverse effect on the Company’s business and could potentially cause the Company to cease operations
Although a substantial amount of the Company’s effort will focus on the continued research and pre-clinical testing, potential approval and commercialization of its existing product candidates, the success of the Company’s business also depends in part upon its ability to identify, license or discover additional product candidates. The Company’s research programs or licensing efforts may fail to yield additional product candidates for clinical development for a number of reasons, including but not limited to the following:
|
● |
the Company’s research or business development methodology or search criteria and process may be unsuccessful in identifying potential product candidates; |
|
● |
the Company may not be able or willing to assemble sufficient resources to acquire or discover additional product candidates; |
|
● |
the Company’s product candidates may not succeed in pre-clinical or clinical testing; |
|
● |
the Company’s product candidates may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval; |
|
● |
competitors may develop alternatives that render the Company’s product candidates obsolete or less attractive; |
|
● |
product candidates the Company develops may be covered by third parties’ patents or other exclusive rights; |
|
● |
the market for a product candidate may change during the Company’s program so that such a product may become unreasonable to continue to develop; |
|
● |
a product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; and |
|
● |
a product candidate may not be accepted as safe and effective by patients, the medical community or third-party payors. |
If any of these events occurs, the Company may be forced to abandon its development efforts to identify, license or discover additional product candidates, which would have a material adverse effect on the Company’s business and could potentially cause the Company to cease operations. Research programs to identify new product candidates require substantial technical, financial and human resources. The Company may focus its efforts and resources on potential programs or product candidates that ultimately prove to be unsuccessful.
There is no assurance that the Company will turn a profit or generate immediate revenues
There is no assurance as to whether the Company will be profitable, earn revenues, or pay dividends. The Company has incurred and anticipates that it will continue to incur substantial expenses relating to the development and initial operations of its business. The payment and amount of any future dividends will depend upon, among other things, the Company’s results of operations, cash flow, financial condition, and operating and capital requirements. There is no assurance that future dividends will be paid, and, if dividends are paid, there is no assurance with respect to the amount of any such dividends.
The Company has incurred operating losses since inception and has not yet generated positive cash flows from operations. There is also no assurance that the Company will achieve or sustain profitability in the future. The Company’s ability to continue operations is dependent upon its ability to successfully execute its business plan, and to secure additional financing as required.
As at September 30, 2025, the Company had cash and cash equivalents and access to capital resources that management believes are sufficient to fund planned operations and capital requirements for at least the next twelve to eighteen months. However, the Company may require additional capital. If the Company is unable to obtain additional financing, this could materially adversely affect the Company’s financial condition, results of operations, and ability to continue operations
The Company may not be able to adequately protect and maintain its intellectual property and licenses, which could result in a material adverse effect to the Company’s business, financial condition and results of operations
The Company’s success will depend in part on its ability to protect and maintain its intellectual property rights and its licenses. No assurance can be given that the license or rights used by the Company will not be challenged, invalidated, infringed or circumvented, nor that the rights granted thereunder will provide competitive advantages to the Company. It is not clear whether the Company’s pending patent applications will result in the issuance of patents. There is no assurance that the Company will be able to enter into licensing arrangements, develop or obtain alternative technology in respect of patents issued to third parties that incidentally cover its production processes. Moreover, the Company could potentially incur substantial legal costs in defending legal actions which allege patent infringement or by instituting patent infringement suits against others. The Company’s commercial success also depends on the Company not infringing patents or proprietary rights of others and not breaching the exclusive license granted to the Company. There can be no assurance that the Company will be able to maintain such licenses that it may require to conduct its business or that such licenses have been obtained at a reasonable cost. Furthermore, there can be no assurance that the Company will be able to remain in compliance with its licenses. Consequently, there may be a risk that such licenses may be withdrawn with no compensation or penalties to the Company.
The Company’s inability to achieve timelines for publicly disclosed projects may result in material adverse effects on the Company’s business, financial condition and results of operations
The Company’s business is dependent on a number of key inputs and their related costs including raw materials and supplies related to its operations, as well as electricity, water and other utilities. Any significant interruption or negative change in the availability or economics of the supply chain for key inputs could materially impact the business, financial condition operating results, and timelines for project development of the Company. Any inability to secure required supplies and services or to do so on appropriate terms could have a materially adverse impact on the business, financial condition, operating results, and timelines for project development of the Company.
The Company may need additional capital for future operations and the Company is unable to secure any required capital it may be forced to curtail or discontinue operations
It is possible that costs associated with the operating the Company’s business will exceed the Company’s projections depending on the timing of future operating and capital expenses. Assuming the Company’s existing funds sustain the Company’s operations for next 12 months, the Company believes that it may thereafter require additional capital for additional product development, sales and marketing operations, other operating expenses and for general corporate purposes to fund growth in our Company’s markets. The Company does not know how much additional funding it may require. The Company may therefore be required to seek other sources of financing in the future, which sources (assuming the Company is able to locate such alternative sources of financing) may be on terms less favorable to the Company than those of our previous securities offerings. Any additional equity financing may be dilutive to shareholders, and debt financing, if available, may involve restrictive covenants. If additional funds are raised through the issuance of equity securities, the percentage ownership of shareholders will be reduced, shareholders may experience additional dilution in net book value per share, or such equity securities may have rights, preferences or privileges senior to those of the holders of the Common Shares. If adequate funds are not available on acceptable terms, the Company may be unable to develop or enhance its products, take advantage of future opportunities or respond to competitive pressures, any of which could have a material adverse effect on the Company’s business, financial condition and operating results, or the Company may be forced to curtail or cease its operations.
The Company may become subject to product liability claims, which could harm the Company’s financial condition and liquidity if the Company is unable to successfully defend or insure against such claims
The risk of product liability is inherent in the research, development, manufacturing, marketing and use of pharmaceutical products. Product candidates and products that the Company may commercially market in the future may cause, or may appear to have caused, injury or dangerous drug reactions, and expose the Company to product liability claims. These claims might be made by patients who use the product, healthcare providers, pharmaceutical companies, corporate collaborators or others selling such products. If the Company’s product candidates during clinical trials were to cause adverse side effects, the Company may be exposed to substantial liabilities. Regardless of the merits or eventual outcome, product liability claims, or other claims related to the Company’s product candidates may result in:
|
● |
decreased demand for the Company’s products due to negative public perception; |
|
● |
damage to the Company’s reputation; |
|
● |
withdrawal of clinical trial participants or difficulties in recruiting new trial participants; |
|
● |
initiation of investigations by regulators; |
|
● |
costs to defend or settle related litigation; |
|
● |
a diversion of management’s time and resources; |
|
● |
substantial monetary awards to trial participants or patients; |
|
● |
product recalls, withdrawals or labeling, marketing or promotional restrictions; |
|
● |
loss of revenues from product sales; and |
|
● |
the inability to commercialize any product candidates, if approved. |
The Company has obtained insurance for all ongoing clinical trials and will continue to obtain such insurance for future clinical trials. However, the insurance coverage may not be sufficient to reimburse the Company for any expenses or losses it may suffer. Insurance coverage is becoming increasingly expensive, and, in the future, the Company, or any of its collaborators, may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts or at all to protect against losses due to liability. Even if the Company’s agreements with any future collaborators entitle it to indemnification against product liability losses, such indemnification may not be available or adequate should any claim arise. The Company’s inability to obtain sufficient product liability insurance at an acceptable cost to protect against product liability claims could prevent or inhibit the commercialization of its product candidates. If a successful product liability claim or series of claims is brought against the Company for uninsured liabilities or in excess of insured liabilities, its assets may not be sufficient to cover such claims and its business operations could be impaired.
Should any of the events described above occur, this could have a material adverse effect on the Company’s business, financial condition and results of operations.
Health and safety issues related to the Company’s products may have a material adverse effect on the Company’s business and results of operations
Health and safety issues related to the Company’s products may arise that could lead to litigation or other action against the Company or to regulation of certain of the Company’s product components. The Company may be required to modify its products and may also be required to pay damages that may reduce the Company’s profitability and adversely affect its financial condition. Even if these concerns prove to be baseless, the resulting negative publicity could affect the Company’s ability to market certain of its products and, in turn, could harm the Company’s business and results from operations.
The Company has international operations, which subjects the Company to risks inherent with operations outside of Canada
The Company has international operations and may seek to obtain market approvals in foreign markets that it deems could generate significant opportunities. However, even with the cooperation of a commercialization partner, conducting drug development in foreign countries involves inherent risks, including, but not limited to: difficulties in staffing, funding and managing foreign operations; different and unexpected changes in regulatory requirements; export restrictions; tariffs and other trade barriers; different reimbursement systems; economic weaknesses or political instability in particular foreign economies and markets; compliance with tax, employment, immigration and labour laws for employees living or travelling abroad; supply chain and raw materials management; difficulties in protecting, acquiring, enforcing and litigating intellectual property rights; fluctuations in currency exchange rates; and potentially adverse tax consequences.
If the Company were to experience any of the difficulties listed above, or any other difficulties, its international development activities and its overall financial condition may suffer and cause it to reduce or discontinue its international development and market approval efforts.
Changes in U.S. and international trade policies may adversely impact the Company’s business and operating results
The U.S. government has made statements and taken actions that have led to certain changes and may lead to additional changes to U.S. and international trade policies. For example, President Trump has imposed or signaled to impose a series of tariffs on certain products manufactured outside the United States, including pharmaceutical products and raw materials and components for pharmaceutical products, and it is unknown whether and to what extent additional tariffs (or other new laws or regulations) will be adopted, or the effect that any such actions would have on the Company or its industry. Any unfavorable government policies on international trade, such as export controls, capital controls or tariffs, may affect the demand for the Company’s product candidates, the competitive position of the Company’s product candidates, and import or export of raw materials and product used in the Company’s drug development, clinical manufacturing and future commercial activities. If any new tariffs, export controls, legislation and/or regulations are implemented, or if existing trade agreements are renegotiated or if the U.S. government takes retaliatory trade actions due to the ongoing trade tensions, such changes could have an adverse effect on the Company’s our business, financial condition and results of operations.
Proposed legislation in the U.S. Congress, including changes in U.S. tax law, may adversely impact certain investors, our business and the value of the Company’s securities
Changes to U.S. tax laws (which changes may have retroactive application) could adversely affect the Company’s business or holders of the Company’s securities. In recent years, many changes to U.S. federal income tax laws have been proposed and made, and additional changes to U.S. federal income tax laws are likely to continue to occur in the future.
The U.S. Congress is currently considering numerous items of legislation which may be enacted prospectively or with retroactive effect, which legislation could adversely impact certain investors, the Company’s financial performance and the value of the Company’s securities. On July 4, 2025, the President of the United States signed into law a new tax bill commonly referred to as the “One Big Beautiful Bill Act”, which may affect the U.S. federal income tax considerations applicable to certain investors of the Company’s securities. The likelihood of other similar legislation being enacted is uncertain, and the provisions of such bill or other similar legislation may change prior to enactment. Investors should consult their own tax advisors regarding such enacted or proposed legislation as well as the potential impact of such legislation on them in light of their own personal circumstances.
Exchange rate fluctuations between the U.S. dollar and the Canadian dollar may negatively affect the Company’s earnings and cash flows
The Company has Canadian, U.S. dollar, and Australia bank accounts and regularly incurs expenses primarily in Australian dollars, Canadian dollars and U.S. dollars. As a result, the Company is exposed to the risks that the Canadian or Australian dollar may devalue relative to the U.S. Dollar, or, if the Canadian or Australian dollar appreciates relative to the U.S. Dollar, that the inflation rate in Canada or Australia may exceed such rate of devaluation of the U.S. dollar, or that the timing of such devaluation may lag behind inflation in Canada. The Company cannot predict any future trends in the rate of inflation in Canada or the rate of devaluation, if any, of the Canadian or Australian dollar against the U.S. Dollar, or similarly of the U.S. Dollar against the Canadian or Australian Dollar. The value of the Company’s holdings in each currency may change over time, shifting the risk from one currency or the other, and the Company may not be able to accurately predict economic or global trends that may impact the Canadian dollar, Australian dollar or the U.S. dollar. The Company, further, may need to operate in other currencies due to the international nature of our operations, and is further exposed to currency risk as may relate to other foreign countries.
If patent laws or the interpretation of patent laws change, the Company’s competitors may be able to develop and commercialize the Company’s discoveries
Important legal issues remain to be resolved as to the extent and scope of available patent protection for biopharmaceutical products and processes in Canada and other important markets outside Canada, such as Europe or the United States. As such, litigation or administrative proceedings may be necessary to determine the validity, scope and ownership of certain of the Company’s and others’ proprietary rights. Any such litigation or proceeding may result in a significant commitment of resources in the future and could force the Company to do one or more of the following: cease selling or using any of its future products that incorporate a challenged intellectual property, which would adversely affect its revenue; obtain a license or other rights from the holder of the intellectual property right alleged to have been infringed or otherwise violated, which license may not be available on reasonable terms, if at all; and redesign its future products to avoid infringing or violating the intellectual property rights of third parties, which may be time-consuming or impossible to do. In addition, changes in patent laws in Canada and other countries may result in allowing others to use its discoveries or develop and commercialize our products. The Company cannot provide assurance that the patents it obtains will afford it significant commercial protection.
The Company may not be able to enforce its intellectual property rights throughout the world. This risk is exacerbated because the Company expects that one or more of our product candidates will be manufactured and used in a number of foreign countries
The laws of foreign countries may not protect intellectual property rights to the same extent as the laws of Canada. Many companies have encountered significant problems in protecting and defending intellectual property rights in certain foreign jurisdictions. This risk is exacerbated for the Company because it expects that future product candidates could be manufactured and used in a number of foreign countries.
The legal systems of some countries, particularly developing countries, do not favor the enforcement of patents and other intellectual property protection, especially those relating to life sciences. This could make it difficult to stop the infringement or other misappropriation of the Company’s intellectual property rights. For example, several foreign countries have compulsory licensing laws under which a patent owner must grant licenses to third parties. In addition, some countries limit the enforceability of patents against third parties, including government agencies or government contractors. In these countries, patents and trade secrets may provide limited or no benefit.
Most jurisdictions in which the Company intends to apply for patents have patent protection laws similar to those of Canada, but some of them do not. The Company may do business in the future in countries that may not provide the same or similar protection as that provided in Canada. Additionally, due to uncertainty in patent protection law, the Company has not filed applications in many countries where significant markets exist.
Proceedings to enforce patent rights in foreign jurisdictions could result in substantial costs and divert the Company’s efforts and attention from other aspects of its business. Accordingly, efforts to protect intellectual property rights in such countries may be inadequate. In addition, changes in the law and legal decisions by courts in Canada, the U.S., and other foreign countries may affect our ability to obtain adequate protection for the Company’s technology and the enforcement of its intellectual property.
The lack of product for commercialization would have a material adverse effect on the Company’s business, financial condition and results of operations
If the Company cannot successfully develop, manufacture and distribute its products, or if the Company experiences difficulties in the development process, such as capacity constraints, quality control problems or other disruptions, the Company may not be able to develop market-ready commercial products at acceptable costs, which would adversely affect the Company’s ability to effectively enter the market. A failure by the Company to achieve a low-cost structure through economies of scale or improvements in cultivation and manufacturing processes would have a material adverse effect on the Company’s commercialization plans and the Company’s business, prospects, results of operations and financial condition.
Failure to develop new and innovative products may have a material adverse effect on the Company’s business
The Company’s success will depend, in part, on its ability to develop, introduce and market new and innovative products. If there is a shift in consumer demand, the Company must meet such demand through new and innovative products or else the Company’s business will fail. The Company’s ability to develop, market and produce new products is subject to the Company having substantial capital. There is no assurance that the Company will be able to develop new and innovative products or have the capital necessary to develop such products.
The lack of experience of the Company’s management in marketing, selling, and distributing products may have a material adverse effect on the Company’s business and financial condition
The Company’s management’s lack of experience in marketing, selling, and distributing the Company’s products could lead to poor decision-making, which could result in cost-overruns and/or the inability to produce the desired products. Although management of the Company intends to hire experienced and qualified staff, this inexperience could also result in the Company’s inability to consummate revenue contracts or any contracts at all. Any combination of the aforementioned may result in the failure of the Company.
The size of the Company’s target market is difficult to quantify, and investors will be reliant on their own estimates on the accuracy of market data
Due to the industry in which the Company operates in being in a nascent stage with uncertain boundaries, there is a lack of information about comparable companies available for potential investors to review in deciding whether to invest in the Company and, few, if any, established companies whose business model the Company can follow or upon whose success the Company can build. Accordingly, investors will have to rely on their own estimates in deciding about whether to invest in the Company. There can be no assurance that the Company’s estimates are accurate or that the market size is sufficiently large for its business to grow as projected, which may negatively impact its financial results.
The Company will continue to sell Common Shares for cash to fund operations and capital expansion that will dilute current shareholders
There is no guarantee that the Company will be able to achieve its business objectives. The continued development of the Company will require additional financing. The failure to raise such capital could result in the delay or indefinite postponement of current business objectives or the Company going out of business. There can be no assurance that additional capital or other types of financing will be available if needed or that, if available, the terms of such financing will be favourable to the Company.
If additional funds are raised through issuances of equity or convertible debt securities, existing shareholders could suffer significant dilution, and any new equity securities issued could have rights, preferences and privileges superior to those of holders of Common Shares. The Company’s articles permit the issuance of an unlimited number of Common Shares, and shareholders will have no pre-emptive rights in connection with such further issuance. The directors of the Company have discretion to determine the price and the terms of issue of further issuances. In addition, from time to time, the Company may enter into transactions to acquire assets or the shares of other companies. These transactions may be financed wholly or partially with debt, which may temporarily increase the Company’s debt levels above industry standards. Any debt financing secured in the future could involve restrictive covenants relating to capital raising activities and other financial and operational matters, which may make it more difficult for the Company to obtain additional capital and to pursue business opportunities, including potential acquisitions. The Company may require additional financing to fund its operations to the point where it is generating positive cash flows. Negative cash flow may restrict the Company’s ability to pursue its business objectives.
Clinical and pre-clinical drug development is a lengthy, costly process with uncertain outcomes. The results from previous clinical trials and early pre-clinical studies of the Company’s product candidates may not predict future results. The regulatory approval process is lengthy and unpredictable. Inability to obtain the regulatory approval can be harmful for business
Before the Company can begin clinical trials, the Company must submit the results of pre-clinical studies, along with other necessary information such as product candidate chemistry, manufacturing controls, and the Company’s proposed clinical trial protocol, to the FDA or other comparable regulatory authorities as part of an investigational new drug application or similar regulatory filing. To obtain marketing approval from the FDA or other comparable foreign regulatory authorities, the Company must complete pre-clinical development and extensive clinical trials to demonstrate their safety and efficacy.
This process is expensive, can take many years, and its outcome is inherently uncertain. The Company’s clinical trials may not be conducted as planned or completed on schedule, if at all, and failure can occur at any time during the pre-clinical study or clinical trial process. Despite promising pre-clinical or clinical results, any product candidate can unexpectedly fail at any stage of development. Historically, the failure rate for product candidates in drug development is high. Results from pre-clinical studies or early clinical trials may not predict the outcomes of later clinical trials, and interim results of a clinical trial are not necessarily indicative of final results. The Company had previously submitted an investigational new drug application to the FDA but later withdrew it prior to full review. In the withdrawal letter, the FDA mentioned partial clinical hold deficiencies related to the proposed dosing regime. Additionally, product candidates in later stages of clinical trials may fail to demonstrate the desired safety and efficacy characteristics, despite having progressed through pre-clinical studies and clinical trials. The FDA or any foreign regulatory authorities may delay, restrict, or deny approval of the Company’s product candidates, or require additional nonclinical or clinical testing, or even force the Company to abandon a program for various reasons.
The Company’s officers and directors may be engaged in a range of business activities resulting in conflicts of interest, which may have a material adverse effect on the Company’s operations
The Company may be subject to various potential conflicts of interest because some of its officers and directors may be engaged in a range of business activities. In addition, the Company’s executive officers and directors may devote time to their outside business interests, so long as such activities do not materially or adversely interfere with their duties to the Company. In some cases, the Company’s executive officers and directors may have fiduciary obligations associated with these business interests that interfere with their ability to devote time to the Company’s business and affairs and that could adversely affect the Company’s operations. These business interests could require significant time and attention of the Company’s executive officers and directors.
In addition, the Company may become involved in other transactions which conflict with the interests of its directors and officers who may from time-to-time deal with persons, firms, institutions or companies with which the Company may be dealing, or which may be seeking investments similar to those desired by it. The interests of these persons could conflict with those of the Company. In addition, from time to time, these persons may be competing with the Company for available investment opportunities. Conflicts of interest, if any, will be subject to the procedures and remedies provided under applicable laws. If such a conflict of interest arises at a meeting of the Company’s directors, a director who has such a conflict will abstain from voting for or against the approval of such participation or such terms. In accordance with applicable laws, the directors of the Company are required to act honestly, in good faith and in the best interests of the Company.
In certain circumstances, the Company’s reputation could be damaged, which may have a material adverse effect on the Company’s financial performance, financial condition, cash flows and growth prospects
Damage to the Company’s reputation can be the result of the actual or perceived occurrence of any number of events, and could include any negative publicity, whether true or not. The increased usage of social media and other web-based tools used to generate, publish and discuss user-generated content and to connect with other users has made it increasingly easier for individuals and groups to communicate and share opinions and views regarding the Company and its activities, whether true or not. Although the Company believes that it operates in a manner that is respectful to all stakeholders and that it takes care in protecting its image and reputation, the Company does not ultimately have direct control over how it is perceived by others. Reputation loss may result in decreased investor confidence, increased challenges in developing and maintaining industry relations and an impediment to the Company’s overall ability to advance its projects, thereby having a material adverse impact on financial performance, financial condition, cash flows and growth prospects.
The Company has negative operating cash flow
The Company’s business has incurred losses since its inception. Although the Company expects to become profitable, there is no guarantee that will happen, and the Company may never become profitable. To date, the Company has not generated any revenue and the Company has a negative operating cash flow which may continue for the foreseeable future. The Company’s ability to generate revenue and potential to become profitable will depend largely on its ability to manufacture and market its products. There can be no assurance that any such events will occur or that the Company will ever become profitable. Even if the Company does achieve profitability, the Company cannot predict the level of such profitability. If the Company sustains losses over an extended period of time, the Company may be unable to continue its business.
The Company’s forward-looking statements may prove to be inaccurate
Investors should not place undue reliance on forward-looking statements. By their nature, forward-looking statements involve numerous assumptions, known and unknown risks and uncertainties, of both general and specific nature, that could cause actual results to differ materially from those suggested by the forward-looking statements or contribute to the possibility that predictions, forecasts or projections will prove to be materially inaccurate. Additional information on the risks, assumptions and uncertainties can be found in this AIF under the heading “Forward-Looking Information”.
If the Company has a material weakness in its internal controls over financial reporting, investors could lose confidence in the reliability of the Company’s financial statements, which could result in a decrease in the value of the Company’s Common Shares
One or more material weaknesses in the Company’s internal controls over financial reporting could occur or be identified in the future. In addition, because of inherent limitations, the Company’s internal controls over financial reporting may not prevent or detect misstatements, and any projections of any evaluation of effectiveness of internal controls to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with our policies or procedures may deteriorate. If the Company fails to maintain the adequacy of its internal controls, including any failure or difficulty in implementing required new or improved controls, the Company’s business and results of operations could be harmed, the Company may not be able to provide reasonable assurance as to its financial results or meet the Company’s reporting obligations and there could be a material adverse effect on the price of the Company’s Common Shares.
Difficulties with forecasts
The Company must rely largely on its own market research to forecast sales as detailed forecasts are not generally obtainable from other sources at this early stage of the biotechnology industry dedicated to the discovery of serotonergic therapeutics. A failure in the demand for the Company’s products to materialize as a result of competition, technological change or other factors could have a material adverse effect on the Company’s business, results of operations and financial condition.
Risks Related to Our Common Shares
Our executive officers and directors beneficially own approximately 14.33% of the Company’s Common Shares.
As of the date of this AIF, our executive officers and directors beneficially own, in the aggregate, approximately 14.33% of the Company’s Common Shares. As a result, they are able to exercise a significant level of control over all matters requiring shareholder approval, including the election of directors, amendments to our articles and approval of significant corporate transactions. This control could have the effect of delaying or preventing a change of control of our Company or changes in management and will make the approval of certain transactions difficult or impossible without the support of these shareholders.
The continued sale of equity securities will dilute the ownership percentage of our existing shareholders and may decrease the market price for the Company’s Common Shares
The Company’s Notice of Articles (as defined herein) authorizes the issuance of an unlimited number of Common Shares. The Board of Directors has the authority to issue additional shares in the Company’s capital stock to provide additional financing in the future. The issuance of any such Common Shares may result in a reduction of the book value or market price of the Company’s outstanding Common Shares. Given the Company’s lack of revenue, the Company will likely have to issue additional equity securities to obtain working capital it requires in the future. The Company’s efforts to fund its intended business plans will therefore result in dilution to existing shareholders. If the Company does issue any such additional Common Shares, such issuance also will cause a reduction in the proportionate ownership and voting power of all other shareholders. As a result of such dilution, if you acquire Common Shares your proportionate ownership interest and voting power could be decreased. Furthermore, any such issuances could result in a change of control or a reduction in the market price for the Company’s Common Shares.
Additionally, we had 359,950 Options, 77,150 RSUs and 361,765 Warrants outstanding as of September 30, 2025. The exercise price of some of these Options and Warrants is below the Company’s current market price, and you could purchase shares in the market at a price in excess of the exercise price of our outstanding Options or Warrants. If the holders of these Options and Warrants elect to exercise them, your ownership position will be diluted and the per share value of the Common Shares you have or acquire could be diluted as well. As a result, the market value of the Company’s Common Shares could significantly decrease as well.
The market price of the Company’s Common Shares may be volatile and may fluctuate in a way that is disproportionate to the Company’s operating performance
Securities of companies with a small market capitalization have experienced substantial volatility in the past, often based on factors unrelated to the companies’ financial performance or prospects. These factors include macroeconomic developments in North America and globally, as well as market perceptions of the attractiveness of particular industries. Factors unrelated to the Company’s performance that may affect the price of the Common Shares include the following: the extent of analytical coverage available to investors concerning the Company’s business may be limited if investment banks with research capabilities do not follow the Company; lessening in trading volume and general market interest in the Common Shares may affect an investor’s ability to trade significant numbers of Common Shares; the size of the Company’s public float may limit the ability of some institutions to invest in Common Shares; and a substantial decline in the price of the Common Shares that persists for a significant period of time could cause the Common Shares to be delisted from the CSE and/or NASDAQ, further reducing market liquidity. As a result of any of these factors, the market price of the Common Shares at any given point in time may not accurately reflect the Company’s long-term value. Securities class action litigation often has been brought against companies following periods of volatility in the market price of their securities. In the future the Company may be the target of similar litigation. Securities litigation could result in substantial costs and damages and divert management’s attention and resources.
The market price of the Common Shares is affected by many other variables which are not directly related to the Company’s success and are, therefore, not within the Company’s control. Such variables include other developments that affect the breadth of the public market for the Common Shares, the release or expiration of lock-up or other transfer restrictions on the Common Shares, and the attractiveness of alternative investments. The effect of these and other factors on the market price of the Common Shares is expected to make the Common Share price volatile in the future, which may result in losses to investors.
The Company’s stock price is expected to be volatile and will be drastically affected by governmental and regulatory regimes and other factors outside of our control. The Company cannot fully predict the results of its operations expected to take place in the future. The results of these activities will inevitably affect our decisions related to future operations and will likely trigger major changes in the trading price of Common Shares.
Volatility in the Company’s Common Share price may subject the Company to securities litigation
The market for the Company’s Common Shares may have, when compared to seasoned issuers, significant price volatility, and the Company expects that its Common Share price may continue to be more volatile than that of a seasoned issuer for the indefinite future. In the past, plaintiffs have often initiated securities class action litigation against a company following periods of volatility in the market price of its securities. The Company may, in the future, be the target of similar litigation. Securities litigation could result in substantial costs and liabilities and could divert management's attention and resources.
The Company does not intend to pay dividends and there will thus be fewer ways in which you are able to make a gain on your investment
The Company has never paid any cash or stock dividends and the Company does not intend to pay any dividends for the foreseeable future. To the extent that the Company requires additional funding in the future, potential funding sources may prohibit the payment of any dividends. Because the Company does not intend to declare dividends, any gain on your investment will need to result from an appreciation in the price of the Company’s Common Shares. There will therefore be fewer ways in which you are able to make a gain on your investment.
FINRA sales practice requirements may limit your ability to buy and sell the Company’s Common Shares, which could depress the price of the Company’s Common Shares
FINRA rules require broker-dealers to have reasonable grounds for believing that an investment is suitable for a customer before recommending that investment to the customer. Prior to recommending speculative low-priced securities to their non-institutional customers, broker-dealers must make reasonable efforts to obtain information about the customer’s financial status, tax status and investment objectives, among other things. Under interpretations of these rules, FINRA believes that there is a high probability such speculative low-priced securities will not be suitable for at least some customers. Thus, FINRA requirements may make it more difficult for broker-dealers to recommend that their customers buy the Company’s Common Shares, which may limit your ability to buy and sell the Company’s Common Shares, have an adverse effect on the market for the Company’s Common Shares and, thereby, depress their market prices.
The Company is a foreign private issuer within the meaning of the rules under the Exchange Act, and as such the Company is exempt from certain provisions applicable to United States domestic public companies
The Company is a foreign private issuer within the meaning of the rules under the Exchange Act. As such, the Company is exempt from certain provisions applicable to United States domestic public companies. For example:
|
• |
the Company is not required to provide as many Exchange Act reports, or as frequently, as a domestic public company; |
|
• |
for interim reporting the Company is permitted to comply solely with its home country requirements, which are less rigorous than the rules that apply to domestic public companies; |
|
• |
the Company is not required to provide the same level of disclosure on certain issues, such as executive compensation; |
|
• |
the Company is exempt from provisions of Regulation FD aimed at preventing issuers from making selective disclosures of material information; |
|
• |
the Company is not required to comply with the sections of the Exchange Act regulating the solicitation of proxies, consents or authorizations in respect of a security registered under the Exchange Act; and |
|
• |
the Company is not required to comply with Section 16 of the Exchange Act requiring insiders to file public reports of their share ownership and trading activities and establishing insider liability for profits realized from any “short-swing” trading transaction. |
Shareholders may not have access to certain information they may deem important and are accustomed to receiving from U.S. reporting companies.
As an “emerging growth company” under applicable law, the Company will be subject to lessened disclosure requirements. Such reduced disclosure may make the Company’s Common Shares less attractive to investors
For as long as the Company remains an “emerging growth company”, as defined in the JOBS Act, the Company will elect to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” and including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in the Company’s periodic reports, exemptions from the requirements of holding a non-binding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. Because of these lessened regulatory requirements, the Company’s shareholders would be left without information or rights available to shareholders of more mature companies. If some investors find the Company’s Common Shares less attractive as a result, there may be a less active trading market for such securities and their market prices may be more volatile.
The Company incurs significant costs as a result of being a public company, which costs will grow after the Company ceases to qualify as an “emerging growth company”
The Company incurs significant legal, accounting and other expenses as a public company. The Sarbanes-Oxley Act, as well as rules subsequently implemented by the SEC and Nasdaq, impose various requirements on the corporate governance practices of public companies. The Company is an “emerging growth company”, as defined in the JOBS Act, and will remain an emerging growth company until the earlier of : (1) the last day of the fiscal year (a) following the fifth anniversary of the date of the first sale of the Company’s common equity securities pursuant to an effective registration statement under the U.S. Securities Act, (b) in which the Company has total annual gross revenue of at least US$1.235 billion or (c) in which the Company is deemed to be a large accelerated filer, which means the market value of the Common Shares that is held by non-affiliates exceeds US$700 million as of the prior June 30th; and (2) the date on which the Company has issued more than US$1.0 billion in non-convertible debt during the prior three-year period. An emerging growth company may take advantage of specified reduced reporting and other requirements that are otherwise applicable generally to public companies. These provisions include exemption from the auditor attestation requirement under Section 404 of the Sarbanes-Oxley Act in the assessment of the emerging growth company’s internal control over financial reporting and permission to delay adopting new or revised accounting standards until such time as those standards apply to private companies.
Compliance with these rules and regulations increases the Company’s legal and financial compliance costs and makes some corporate activities more time-consuming and more costly. After the Company is no longer an emerging growth company, the Company expects to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements of Section 404 and the other rules and regulations of the SEC. For example, as a public company, the Company has been required to increase the number of independent directors and adopt policies regarding internal controls and disclosure controls and procedures. The Company has incurred additional costs in obtaining director and officer liability insurance. In addition, the Company incurs additional costs associated with its public company reporting requirements. It may also be more difficult for the Company to find qualified persons to serve on its Board of Directors or as executive officers. The Company is currently evaluating and monitoring developments with respect to these rules and regulations, and the Company cannot predict or estimate with any degree of certainty the amount of additional costs it may incur or the timing of such costs.
DIVIDENDS AND DISTRIBUTIONS
The Company has not declared nor paid any cash dividends on any of its issued equity securities since its inception. Other than requirements imposed under applicable corporate law, there are no other restrictions on the Company’s ability to pay dividends under the Company’s constating documents. The Company has not paid any dividends on the Common Shares since its incorporation. The Company has no present intention of paying dividends on the Common Shares, as it anticipates that all available funds will be invested to finance the growth of its business and, when appropriate, retire debt.
DESCRIPTION OF CAPITAL STRUCTURE
Common Shares
The Company’s authorized share capital consists of an unlimited number of Common Shares without par value. As of the date of this AIF, there are 7,787,161 Common Shares issued and outstanding as fully paid and non-assessable.
The holders of the Common Shares are entitled to dividends, if, as and when declared by the Board of Directors, to receive notice of meetings of shareholders of the Company, to one vote per Common Share at meetings of the shareholders of the Company and, upon liquidation, to receive such assets of the Company as are distributable to the holders of the Common Shares. Holders of Common Shares do not have cumulative voting rights with respect to the election of directors and, accordingly, holders of a majority of the votes eligible to vote at a meeting of shareholders may elect all the directors of the Company standing for election. Dividends, if any, will be paid on a pro rata basis only from funds legally available therefore. The Common Shares do not carry any pre-emptive, subscription, redemption or conversion rights, nor do they contain any sinking or purchase fund provisions.
Warrants
The following table sets forth all Warrants of the Company that are outstanding as of the date of this AIF:
|
Expiry Date |
Number of Warrants |
Exercise Price (CAD$) |
|
December 22, 2028 |
361,765 |
1.70 |
|
Total |
361,765 |
Options
The following table discloses all outstanding Options as of the date of this AIF:
|
Expiry Date |
Number of Options |
Exercise Price |
|
February 16, 2028 |
27,250 |
CAD$5.25 |
|
March 22, 2029 |
102,500 |
CAD$1.84 |
|
October 3, 2029 |
55,000 |
CAD$1.65 |
|
February 26, 2030 |
161,000 |
USD$35.00 |
|
October 30, 2030 |
43,000 |
USD$54.47 |
|
Total |
388,750 |
Restricted Share Units
The following table sets forth all RSUs of the Company that are outstanding as of the date of this AIF:
|
Expiry Date |
Number of RSUs |
Share Price (CAD$) |
|
February 1, 2027 |
12,000 |
N/A |
|
April 27, 2027 |
10,000 |
N/A |
|
December 1, 2027 |
55,000 |
N/A |
|
March 3, 2030 |
150 |
N/A |
|
Total |
77,150 |
MARKET FOR SECURITIES
Trading Price and Volume
On February 8, 2021, the Company’s Common Shares began trading on the CSE under the trading symbol “DRUG”. On November 8, 2021, the Company’s Common Shares began trading on the Nasdaq under the trading symbol “DRUG”.
The tables below summarizes the price ranges and trading volume of Common Shares on the CSE and the NASDAQ for each of the months stated:
CSE Trading Data
|
Month |
Price Range (CDN$) |
Volume |
|||||||
|
High |
Low |
||||||||
|
December 22, 2025 |
$131.88 |
$96.82 | 30,147 | ||||||
|
November 2025 |
$102.45 | $71.85 | 16,171 | ||||||
|
October 2025 |
96.50 | 70.11 | 10,650 | ||||||
|
September 2025 |
84.39 | 53.99 | 7,770 | ||||||
|
August 2025 |
61.48 | 44.69 | 9,898 | ||||||
|
July 2025 |
49.94 | 33.32 | 16,763 | ||||||
|
June 2025 |
40 | 33.01 | 5,562 | ||||||
|
May 2025 |
50.74 | 35.25 | 7,714 | ||||||
|
April 2025 |
53.48 | 38.84 | 11,860 | ||||||
|
March 2025 |
53.74 | 44.86 | 11,802 | ||||||
|
February 2025 |
66.76 | 47.82 | 16,057 | ||||||
|
January 2025 |
61 | 40.65 | 39,215 | ||||||
|
December 2024 |
67.53 | 49.26 | 34,044 | ||||||
|
November 2024 |
79.98 | 42.35 | 161,896 | ||||||
|
October 2024 |
108 | 1.35 | 789,168 | ||||||
NASDAQ Trading Data
|
Month |
Price Range (USD$) |
Volume |
|||||||
|
High |
Low |
||||||||
|
December 22, 2025 |
$97.75 |
$68.43 | 2,821,306 | ||||||
|
November 2025 |
$71.49 | $50.00 | 3,272,300 | ||||||
|
October 2025 |
$70.23 | $48.94 | 3,273,612 | ||||||
|
September 2025 |
$61.72 | $38.00 | 2,197,566 | ||||||
|
August 2025 |
$46.13 | $31.98 | 1,070,393 | ||||||
|
July 2025 |
$37.59 | $24.00 | 848,052 | ||||||
|
June 2025 |
$29.35 | $23.18 | 1,075,244 | ||||||
|
May 2025 |
$37.00 | $25.30 | 602,007 | ||||||
|
April 2025 |
$37.52 | $26.96 | 640,306 | ||||||
|
March 2025 |
$38.33 | $31.05 | 823,824 | ||||||
|
February 2025 |
$47.50 | $32.51 | 1,200,144 | ||||||
|
January 2025 |
$42.47 | $28.21 | 1,867,687 | ||||||
|
December 2024 |
$49.46 | $34.29 | 2,259,452 | ||||||
|
November 2024 |
$58.00 | $30.67 | 5,626,332 | ||||||
|
October 2024 |
$79.02 | $0.94 | 185,507,952 | ||||||
Prior Sales
During the fiscal year ended September 30, 2025 and to the date of this AIF, the Company issued the following securities that are not listed or quoted on a marketplace:
|
Date Issued |
Type of Security |
Number of Securities Issued |
Exercise Price |
|
October 30, 2025 |
Options |
43,000 |
US$54.47 |
|
March 3, 2025 |
RSUs |
600 |
N/A |
|
October 3, 2024 |
Options |
70,000 |
$1.65 |
| February 26, 2025 | Options | 161,000 | US$35.00 |
ESCROWED SECURITIES AND SECURITIES SUBJECT TO CONTRACTUAL RESTRICTIONS ON TRANSFER
As of the date of this AIF, no securities of the Company are currently held in escrow.
DIRECTORS AND OFFICERS
Name, Occupation and Security Holding
The following table sets forth information regarding the directors and executive officers of the Company as of the date of this AIF. The information as to the principal occupation, business or employment is not within the knowledge of the Company and has been furnished by the respective director/officer. The Board has four committees, each comprised of a majority of independent directors: an audit committee, a nominating and corporate governance committee, a compensation committee, and a corporate disclosure committee.
|
Name, province and country of residence |
Position with the Company |
Principal Occupation During the Past Five Years |
Period as Director and/or Officer |
Number of Common Shares and Percentage of Common Shares Held(1) |
|
Ian McDonald, Dubai, UAE |
President, Chief Executive Officer, and Director |
See biographies below |
May 31, 2019 – Present |
1,004,900 (12.90%) |
|
Ryan Cheung British Columbia, Canada |
Chief Financial Officer and Corporate Secretary |
See biographies below |
May 29, 2020 - Present |
Nil (0%) |
|
Stephen D. Collins Illinois, USA |
Chief Medical Officer |
See biographies below |
January 7, 2025 - Present |
Nil (0%) |
|
Alex Vasilkevich Illinois, USA |
Chief Operating Officer, Senior Scientific Officer |
See biographies below |
June 2024 – Present |
27,750 (0.36%) |
|
Nils Christian Bottler(2)(3)(4)(5) Berlin, Germany |
Director |
See biographies below |
September 29, 2020 - Present |
5,000 (0.06%) |
|
Jeremy Fryzuk(2)(3)(4)(5) London, U.K. |
Director |
See biographies below |
September 29, 2020 - Present |
20,609 (0.26%) |
|
Jan Pedersen Region Hovedstaden, Denmark |
Chief Scientific Officer and Director |
See biographies below |
April 27, 2022 - Present |
57,600 (0.74%) |
|
David Weiner(2)(3)(4)(5) New York, USA |
Director |
See biographies below |
February 16, 2023 |
Nil (0%) |
| Notes: |
|
|
(1) |
Percentage is based on 7,787,161 Common Shares issued and outstanding as of the date of this AIF. |
|
(2) |
Member of the Audit Committee. |
|
(3) |
Member of the Nominating and Corporate Governance Committee |
|
(4) |
Member of the Compensation Committee |
|
(5) |
Member of the Corporate Disclosure Committee |
Term of Office
The term of office of each director of the Company expires at the end of the next annual meeting of Shareholders.
Director and Officer Share Ownership
As of the date of the AIF, the Company’s directors and executive officers, as a group, beneficially owned, directly or indirectly, or exercised control or direction over 1,115,859 Common Shares, representing approximately 14.33% of the issued and outstanding Common Shares.
Biographies
The following are brief profiles of the executive officers and directors of the Company.
Ian McDonald, President, Chief Executive Officer and Director - Age: 38
Mr. McDonald is an entrepreneur and former Investment Banker. Prior to joining the Company, Mr. McDonald served on the management team at a TSX-listed gold mining company. In that capacity, Mr. McDonald developed and implemented the corporate strategy as it relates to M&A and capital markets resulting in a $160 million sale within one year. Previously, he worked in a senior role at a Canadian Investment Bank and in private equity in Vancouver, London and Toronto. Under Mr. McDonald’s guidance, clients raised hundreds of millions of dollars in capital. Mr. McDonald has served as a member of the Board of Directors of several TSX Venture Exchange, Canadian Securities Exchange listed and private companies.
Ryan Cheung, Chief Financial Officer - Age: 47
Mr. Cheung is the founder and managing partner of MCPA Services Inc., Chartered Professional Accountants, in Vancouver, B.C. Leveraging his experience as a former auditor of junior venture and resource companies, Mr. Cheung serves as a director and officer or consultant for public and private companies, providing financial reporting, taxation and strategic guidance.
He has been an active member of the Chartered Professional Accountants of British Columbia (formerly Institute of Chartered Accountants of British Columbia) since January 2008. Mr. Cheung holds a diploma in accounting from the University of British Columbia and a Bachelor of Commerce in international business from the University of Victoria.
Stephen D. Collins, Chief Medical Officer - Age: 73
Dr. Collins previously served as CEO of Biscayne Neurotherapeutics, a small molecule clinical stage company focused on CNS disorders, and CEO and President of Biscayne Pharmaceuticals. Biscayne was sold to Supernus Pharmaceuticals in October of 2018 for future development. Before heading the two Biscayne companies, Dr. Collins held several top leadership positions at companies targeting CNS disorders, including as CEO of Neurotherapeutics Pharma and Chief Scientific Officer & VP for Clinical Affairs of Ovation Pharmaceuticals, a CNS-focused biopharmaceutical company acquired by Lundbeck A/S for US $963 million.
Prior to Ovation, Steve served as a Global Director at Johnson & Johnson, overseeing early-stage development of a variety of CNS agents and as a member of the global in-licensing advisory team. Prior to Johnson and Johnson, he worked in Abbott Laboratories’ Pharmaceutical and Hospital Products Divisions, where he developed drugs for a range of CNS indications, including Depakote, Depakote ER, Depacon, and Gabitril, as well as a range of older seizure medicines. Dr. Collins was responsible for all preclinical and clinical programs supporting the successful approval of multiple NDA and sNDA submissions. Dr. Collins has served on several Boards of companies that have developed epilepsy drugs, including Spinifex Pharmaceuticals, which was acquired by Novartis, and Engage Therapeutics, which was sold to UCB S.A.
Dr. Collins has served on the faculty of medicine at Case Western Reserve University and the University of California-San Francisco and was principal investigator or investigator on over 40 drug and device trials. Dr. Collins earned his MD and PhD at Case Western Reserve University after completing undergraduate studies in Biophysics at the University of California, Berkeley.
Alex Vasilkevich, Chief Operating Officer, Senior Strategic Officer - Age: 37
Mr. Vasilkevich, MSc, is an experienced biotech entrepreneur, executive and scientific leader focused on the development of novel serotonergic therapeutics for CNS indications, including epilepsy and neuropsychiatric disorders. He currently serves as Chief Operating Officer at the Company, where he oversees both strategic operations and R&D activities. Mr. Vasilkevich has over 15 years of experience in R&D of pharma and food products. Prior to joining the Company, he managed a R&D portfolio at Lamyra‑Pharmacare, where he supervised the formulation of more than 30 products, established regulatory activities and led business development activities across Europe and Asia. He also served as a consultant for early-stage biotech startups. Mr Vasilkevich has MSc in biotechnology obtained in the Academy of Sciences of Belarus.
Nils Christian Bottler, Director - Age: 38
Mr. Bottler is a venture capitalist currently working at Think.Health Ventures as an associate partner. The company focuses on investment in early-stage start-ups in the fields of digital health and medical device technology. Think.Health supports its portfolio beyond financial investment with knowledge, experience and access to an extensive business network. Mr. Bottler’s prior work experience was in the banking industry working mainly on M&A projects as well as on a number of consulting projects in Germany, China, the UK, and the United Arab Emirates. He then moved to digital media and analyzed, developed and executed new business models at the Axel Springer SE in Berlin before taking a deep dive into the German health care market as SVP RHÖN-Innovations and the premier hospital chain RHÖNKLINIKUM AG.
Jeremy Fryzuk, Director - Age: 40
Mr. Fryzuk is a private equity investment professional based in London. He has over 10 years of experience in private equity. He started his career in investment banking in Toronto with BMO Capital Markets. Mr. Fryzuk holds a Bachelor of Commerce with a major in Finance from Dalhousie University in Canada.
Jan Pedersen, Director and Chief Science Officer - Age: 61
Jan Pedersen, PhD, MSc, is an innovative and highly experienced leader in drug discovery research, with more than 25 years of expertise in neuroscience research management. Dr. Pedersen’s academic interests include neurodegeneration, bioinformatics, biophysics and drug discovery R&D. He is the founder of Torleif Science ApS, a consultancy company aimed at delivering innovation and new ideas in neuroscience. Prior to that, Dr. Pedersen spent 20 years at Lundbeck, a global pharmaceutical company specialized in brain diseases, in positions of increasing responsibility, including building its neurodegeneration/Alzheimer’s disease pipeline, and bringing research programs to the clinic. Dr. Pedersen received an MSc in Chemistry from DTU – Technical University of Denmark, and a PhD in biophysics from the University of Bath.
David Weiner, Director - Age 61
Dr. Weiner has over 25 years of experience in the discovery and clinical development of novel therapeutics for neurological, psychiatric and rare diseases. He began his career at ACADIA Pharmaceuticals, where he held a series of discovery research and clinical development roles working on multiple central nervous system (CNS) therapeutics, most notably pimavanserin, a 5-HT2A receptor inverse agonist, which is approved for the treatment of Parkinson’s disease psychosis. Dr. Weiner also served as the Chief Medical Officer (CMO) and Interim Chief Executive Officer (CEO) for Proteostasis Therapeutics, CMO at aTyr Pharma and Lumos Pharma, CEO at Amathus Therapeutics, and as an independent board member and senior executive at Eleusis, a company focused on therapeutic development of psychedelics and novel 5-HT2A receptor agonists. He has authored more than 30 scientific publications and patents and serves on multiple clinical and scientific advisory boards, including the Michael J. Fox Foundation for Parkinsons Research. He received his M.D. from the School of Medicine and Biomedical Sciences, SUNY at Buffalo, was a Howard Hughes Medical Institute Research Scholar at the NIH, trained in neurology at New York Hospital, Memorial Sloan Kettering, Cornell Medical Center, and did a post-doctoral fellowship in neuropharmacology at the University of Vermont.
Cease Trade Orders, Bankruptcies, Penalties or Sanctions
Other than as disclosed below, no director or executive officer of the Company is, as at the date of this AIF, or has been within 10 years before the date of this AIF, a director, chief executive officer or chief financial officer of any company (including the Company), that:
|
(a) |
was subject to a cease trade order, an order similar to a cease trade order, or an order that denied the relevant company access to any exemption under securities legislation, that was in effect for a period of more than 30 consecutive days, that was issued while the director or executive officer was acting in the capacity as director, chief executive officer or chief financial officer, or |
|
(b) |
was subject to a cease trade order, an order similar to a cease trade order, or an order that denied the relevant company access to any exemption under securities legislation, that was in effect for a period of more than 30 consecutive days, that was issued after the director or executive officer ceased to be a director, chief executive officer or chief financial officer and which resulted from an event that occurred while that person was acting in the capacity as director, chief executive officer or chief financial officer. |
Mr. Ryan Cheung is currently the CFO of DMG, a company listed on the TSXV. DMG was issued a failure-to-file cease trade order on February 1, 2019 by the BCSC for failing to file its annual audited financial statements for the year ended September 30, 2018 and the related management’s discussion and analysis and certification. This failure-to-file cease trade order was revoked on August 28, 2019.
No director or executive officer of the Company, nor a shareholder holding a sufficient number of securities of the Company to affect materially the control of the Company:
|
(a) |
is, as at the date of this AIF, or has been within 10 years before the date of this AIF, a director or executive officer of any company (including the Company) that, while that person was acting in that capacity, or within a year of that person ceasing to act in that capacity, became bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency or was subject to or instituted any proceedings, arrangement or compromise with creditors or had a receiver, receiver manager or trustee appointed to hold its assets; or |
|
(b) |
has, within 10 years before the date of this AIF, become bankrupt, made a proposal under any legislation relating to bankruptcy or insolvency, or become subject to or instituted any proceedings, arrangement or compromise with creditors, or had a receiver, receiver manager or trustee appointed to hold the assets of the proposed director. |
No director or executive officer of the Company has been subject to:
|
(a) |
any penalties or sanctions imposed by a court relating to securities legislation or by a securities regulatory authority or has entered into a settlement agreement with a securities regulatory authority; or |
|
(b) |
any other penalties or sanctions imposed by a court or regulatory body that would likely be considered important to a reasonable security holder in deciding whether to vote for a proposed director. |
Conflicts of Interest
The Company’s directors and officers may serve as directors or officers, or may be associated with, other reporting companies, or have significant shareholdings in other public companies. To the extent that such other companies may participate in business or asset acquisitions, dispositions, or ventures in which the Company may participate, the directors and officers of the Company may have a conflict of interest in negotiating and concluding terms respecting the transaction. If a conflict of interest arises, the Company will follow the provisions of the BCBCA dealing with conflict of interest. These provisions state that where a director has such a conflict, that director must, at a meeting of the Company’s directors, disclose his or her interest and refrain from voting on the matter unless otherwise permitted by the BCBCA. In accordance with the laws of the Province of British Columbia, the directors and officers of the Company are required to act honestly, in good faith, and the best interest of the Company.
LEGAL PROCEEDINGS AND REGULATORY ACTIONS
Other than as disclosed below, there are no legal proceedings or regulatory actions to which the Company is or was a party to or of which any of its property is or was the subject of during the year ended September 30, 2025, or in the subsequent months to the date of this AIF and the Company is not aware of any such proceedings that are pending, threatened or contemplated.
On April 14, 2023, Revati Inc., the consulting company of the Company’s former Chief Medical Officer, Dr. Revati Shreeniwas, commenced legal proceedings against the Company in the Supreme Court of British Columbia in connection with the termination of Revati Inc.’s consulting services. The plaintiff is seeking damages for alleged breach of contract, including:
|
● |
consulting fees equivalent to three months from the date of termination; |
|
● |
the value of certain RSUs and Options that the plaintiff asserts should have vested; |
|
● |
damages for outstanding fees, bad faith, punitive damages, and other related amounts; and |
|
● |
reimbursement for certain expenses, together with pre- and post-judgment interest and legal costs. |
A response to civil claim was filed by the Company on May 9, 2023, opposing all relief sought by the plaintiff. The proceeding is scheduled for trial in the Supreme Court of British Columbia commencing August 4, 2026. The Company disputes both the basis and financial amount of the claims.
PROMOTERS
To the knowledge of the Company’s board, management is not aware of any person or company who could currently be or would have been within the two (2) years immediately preceding the date of this Annual Information Form characterized as a promoter for the Company or a subsidiary of the Company.
INTERESTS OF MANAGEMENT AND OTHERS IN MATERIAL TRANSACTIONS
Other than as disclosed below, elsewhere in this AIF and in the audited consolidated financial statements of the Company for the year ended September 30, 2025, to the best of the Company’s knowledge, none of the directors or executive officers of the Company, or any Shareholders who beneficially own, control or direct, directly or indirectly, more than 10% of the Company’s outstanding Common Shares, or any known associates or affiliates of such persons, had any material interests, direct or indirect, in any transaction within the three most recently completed fiscal years or during the current fiscal year that has materially affected or is reasonably expected to materially affect the Company.
As disclosed under GENERAL DEVELOPMENT OF THE BUSINESS - Three-Year History- Financial Year Ended September 30, 2024, Ian McDonald, the CEO and a director of the Company, was the sole subscriber in the December 2023 Private Placement. Mr. McDonald’s participation in the December 2023 Private Placement allowed the Company to finance and accelerate its Phase 2 Clinical Trial for BMB-101 in a pediatric epilepsy indication.
AUDITOR, TRANSFER AGENT AND REGISTRAR
Auditor
The Company’s auditor is De Visser Gray LLP, Chartered Professional Accountants, of 401 – 905 West Pender Street, Vancouver, British Columbia. De Visser Gray LLP has served as the Company’s auditor since October 19, 2020.
Transfer Agent and Registrar
The registrar and transfer agent of the Company is Computershare Investor Services Inc. at its offices in Vancouver, British Columbia.
MATERIAL CONTRACTS
Other than the contracts listed below and as entered into in the ordinary course of business, the Company has not entered into any material contracts not disclosed elsewhere in this AIF.
|
● |
The Equity Distribution Agreement. |
|
● |
The Exclusive License Agreement. |
AUDIT COMMITTEE
Under National Instrument 52-110 - Audit Committees (“NI 52-110”), a reporting issuer is required to provide disclosure annually with respect to its Audit Committee, including the text of its audit committee charter, information regarding composition of the audit committee, and information regarding fees paid to its external auditor. The Company provides the following disclosure with respect to its Audit Committee.
Audit Committee Charter
The Audit Committee has a charter, a copy of which is attached as Schedule “A” hereto.
Composition of the Audit Committee
The Audit Committee is comprised of the following individuals:
|
Member |
Independence(1) |
Financial Literacy |
|
Nils Bottler(2) |
Independent |
Financially Literate |
|
Jeremy Fryzuk |
Independent |
Financially Literate |
|
David Weiner |
Independent |
Financially Literate |
Notes:
|
(1) |
Within the meaning of NI 52-110. |
|
(2) |
Chair of Audit Committee. |
An Audit Committee member is independent if the member has no direct or indirect material relationship with the Company that could, in the view of the Board, reasonably interfere with the exercise of a member’s independent judgment.
An Audit Committee member is financially literate if they have the ability to read and understand a set of financial statements that present a breadth of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Company’s financial statements.
Relevant Education and Experience
Each member of the Audit Committee has adequate education and experience relevant to their performance as an Audit Committee member and, in particular, the requisite education and experience that provides the member with:
|
(a) |
an understanding of the accounting principles used by the Company to prepare its financial statements and the ability to assess the general application of those principles in connection with estimates, accruals and reserves; |
|
(b) |
experience preparing, auditing, analyzing or evaluating financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of issues that can reasonably be expected to be raised by the Company’s financial statements or experience actively supervising individuals engaged in such activities; and |
|
(c) |
an understanding of internal controls and procedures for financial reporting. |
See “DIRECTORS AND OFFICERS” above, in particular the biographies of each Audit Committee member, for more information concerning each Audit Committee member’s education and experience.
Audit Committee Oversight
The Audit Committee has not made any recommendations to the Board to nominate or compensate any auditor other than De Visser Gray LLP, Chartered Professional Accountants.
Pre-Approval Policies and Procedures
Formal policies and procedures for the engagement of non-audit services have yet to be formulated and adopted. Subject to the requirements of NI 52-110, the engagement of non-audit services is considered by, as applicable, the Board and the Audit Committee, on a case-by-case basis.
External Auditor Service Fees
The Audit Committee has reviewed the nature and amount of the non-audit services provided by De Visser Gray LLP, Chartered Professional Accountants, to the Company to ensure auditor independence. Payments to De Visser Gray LLP, Chartered Professional Accountants, for audit and non-audit services in the years ended September 30, 2025, and September 30, 2024, are outlined in the following table:
|
Financial Period Ending |
Audit Fees ($)(1) |
Audit Related Fees ($)(2) |
Tax Fees ($)(3) |
All Other Fees ($)(4) |
|
September 30, 2025 |
$71,000 |
Nil |
$2,500 |
Nil |
|
September 30, 2024 |
$60,500 |
Nil |
$2,500 |
Nil |
Notes:
|
(1) |
“Audit Fees” include fees necessary to perform the annual audit and quarterly reviews of the Company’s consolidated financial statements. Audit Fees include fees for review of tax provisions and for accounting consultations on matters reflected in the financial statements. Audit Fees also include audit or other attest services required by legislation or regulation, such as comfort letters, consents, reviews of securities filings and statutory audits. |
|
(2) |
“Audit-Related Fees” include services that are traditionally performed by the auditor. These audit-related services include quarterly financial statement reviews, employee benefit audits, due diligence assistance, accounting consultations on proposed transactions, internal control reviews, and audit or attest services not required by legislation or regulation. |
|
(3) |
“Tax Fees” include fees for all tax services other than those included in “Audit Fees” and “Audit-Related Fees”. This category includes fees for tax compliance, tax planning, and tax advice. Tax planning and tax advice includes assistance with tax audits and appeals, tax advice related to mergers and acquisitions, and requests for rulings or technical advice from tax authorities. |
|
(4) |
“All Other Fees” include all other non-audit services. |
INTERESTS OF EXPERTS
Names of Experts
The Company’s auditors are De Visser Gray LLP, who have prepared an independent auditor’s report in respect of the Company’s audited consolidated annual financial statements for the most recent fiscal year ended September 30, 2025. De Visser Gray LLP has advised that they are independent with respect to the Company within the meaning of the CPABC Code of Professional Conduct.
Interests of Experts
To the knowledge of management of the Company, none of the persons above held, at the time of or after such person prepared the statement, report or valuation, any registered or beneficial interests, direct or indirect, in any securities or other property of the Company or of one of its associates or affiliates or is or is expected to be elected, appointed or employed as a director, officer or employee of the Company or of any associate or affiliate of the Company.
ADDITIONAL INFORMATION
Additional financial information relating to the Company is provided in the Company’s audited consolidated financial statements for the year ended September 30, 2025, and Management’s Discussion and Analysis for the year ended September 30, 2025. Additional information, including directors’ and officers’ remuneration and indebtedness, principal holders of your company’s securities and securities authorized for issuance under equity compensation plans, if applicable, is contained in the Company’s information circular for its most recent annual meeting of securityholders. Copies of the Company’s audited annual financial statements, most current interim financial statements, Management’s Discussion and Analysis, and a copy of this AIF, as well as additional information relating to the Company may be found under the Company’s SEDAR+ profile at www.sedarplus.ca.
SCHEDULE A
BRIGHT MINDS BIOSCIENCES INC.
AUDIT COMMITTEE CHARTER
OBJECTIVES
The Corporation’s Audit Committee (the “Audit Committee”) will assist the Corporation’s Board of Directors (the “Board of Directors”) in fulfilling its oversight responsibilities for:
|
1. |
the system of internal control over financial reporting; |
|
2. |
the audit process; |
|
3. |
compliance with legal and regulatory requirements; and |
|
4. |
the processes for identifying, evaluating and managing the Corporation’s principal risks impacting financial reporting. |
Committee Membership
The Board of Directors shall appoint annually from among its members an Audit Committee to hold office for the ensuing year or until their successors are elected or appointed (each, a “Member”).
The Audit Committee shall be composed of at least three directors, and not more than five directors. All Members must meet the independence and audit committee composition requirements promulgated by all governmental and regulatory bodies having jurisdiction over the Corporation as may be in effect from time to time, including Rule 10A-3 under the Exchange Act, Rule 5605(a)(2) of the Listing Rules of the NASDAQ, National Instrument 52-110 – Audit Committees, and the relevant rules of any other stock exchange(s) on which the Corporation’s securities are listed. In general, each member of the Audit Committee must be free from any relationship that, in the view of the Board of Directors, could be reasonably be expected to interfere with the exercise of their judgement as a Member.
All members of the Audit Committee must be financially literate (which is defined as the ability to read and understand a set of financial statements that present a breadth and level of complexity of accounting issues that are generally comparable to the breadth and complexity of the issues that can reasonably be expected to be raised by the Corporation’s financial statements). At least one member of the Audit Committee must satisfy the definition of “financial expert” as set out in Item 407(d)(5)(ii) of Regulation S-K under the United States Securities Act of 1933, as amended, and the Exchange Act.
The Board of Directors may from time to time designate one of the Members of the Audit Committee to be the Audit Committee Chair (the “Chair”) and, unless otherwise determined by the Board of Directors, the Corporate Secretary of the Corporation shall be the Secretary of the Audit Committee (the “Audit Committee Secretary”).
Any member of the Audit Committee may be removed or replaced at any time by the Board of Directors and will cease to be a Member of the Audit Committee on ceasing to be a director of the Corporation. The Board of Directors may fill vacancies on the Audit Committee by election from among the Board of Directors. If and whenever a vacancy will exist on the Audit Committee, the remaining Members may exercise all powers of the Audit Committee so long as a quorum remains.
No Member of the Audit Committee shall receive, directly or indirectly, other than for service on the Board of Directors, the Audit Committee, or other committees of the Board of Directors, any consulting, advisory, or other compensatory fee from the Corporation or any of its related parties or subsidiaries.
Limitations on Audit Committee’s Duties
In contributing to the Audit Committee’s discharge of its duties, each Member of the Audit Committee will be obliged only to exercise the care, diligence and skill that a reasonably prudent person would exercise in comparable circumstances. Nothing in this Charter is intended or may be construed as imposing on any Member of the Audit Committee a standard of care or diligence that is in any way more onerous or extensive than the standard to which any member of the Board of Directors may be otherwise subject.
Members of the Audit Committee are entitled to rely, absent actual knowledge to the contrary, on: (a) the integrity of the persons and organizations from whom they receive information; (b) the accuracy and completeness of the information provided; (c) representations made by management of the Corporation (“Management”) as to the non-audit services provided to the Corporation by the external auditor; (d) financial statements of the Corporation represented to them by a Management or in a written report of the external auditors to present fairly the financial position of the Corporation in accordance with applicable generally accepted accounting principles; and (e) any report of a lawyer, accountant, engineer, appraiser or other person whose profession lends credibility to a statement made by any such person.
Meetings and Participation
The Audit Committee shall meet at least once per quarter, or more frequently as circumstances dictate. The Corporation’s Chief Executive Officer, Chief Financial Officer, any Member of the Audit Committee, or the external auditor may call a meeting of the Audit Committee. The Corporation’s auditors shall be provided notice of all meetings of the Audit Committee and be entitled to attend and be heard thereat.
Meeting agendas will be prepared and provided in advance to Members, along with appropriate briefing materials. The agenda will be set by the Chair in consultation with other Members of the Audit Committee, the Board of Directors and Management of the Corporation.
No business may be transacted by the Audit Committee except at a meeting of its Members at which a quorum of the Audit Committee is present. A quorum for meetings of the Audit Committee is a majority of its Members.
The Audit Committee may ask members of Management and employees of the Corporation (including, for greater certainty, its affiliates and subsidiaries) or others (including the external auditor and legal counsel) to attend meetings and provide such information as the Audit Committee requests. Members of the Audit Committee will have full access to information of the Corporation (including, for greater certainty, its affiliates, subsidiaries and their respective operations) and will be permitted to discuss such information and any other matters relating to the results of operations and financial position of the Corporation with Management, employees, the external auditor and others as they consider appropriate.
The Audit Committee shall keep minutes of its meetings in which shall be recorded all action taken by it, which minutes shall be approved by Audit Committee Members and available as soon as possible to the Board of Directors.
The Audit Committee or its Chair should meet at least once per year with Management and the external auditor in separate sessions to discuss any matters that the Audit Committee or either of these groups desires to discuss privately. In addition, the Audit Committee or its Chair should meet with Management quarterly in connection with the Corporation’s interim financial statements.
Duties, Powers, and Responsibilities
The Audit Committee is hereby delegated the following duties and powers, without limiting these duties and powers, the Audit Committee shall:
|
1. |
Financial Reporting |
|
(a) |
Ensure, through discussions with Management and the external auditors, that the Corporation’s annual and quarterly financial statements (individually and collectively, the “Financial Statements”), as applicable, present fairly in all material respects the financial condition, results of operations and cash flows of the Corporation as of and for the periods presented. |
|
(b) |
Review and recommend for approval to the Board of Directors the Corporation’s Financial Statements, accounting policies that affect the Financial Statements, annual MD&A and associated press release(s). |
|
(c) |
Review the financial statements and other financial information of material subsidiaries of the Corporation and any auditor recommendations concerning such subsidiaries. |
|
(d) |
Be satisfied as to the adequacy of procedures in place for the review of the Corporation’s public disclosure of financial information extracted or derived from annual or quarterly Financial Statements and periodically assess the adequacy of such procedures. |
|
(e) |
Review and approve quarterly Financial Statements, accounting policies that affect the Financial Statements, the quarterly MD&A and the associated press release(s). |
|
(f) |
In review of the annual and quarterly Financial Statements, discuss the quality of the Corporation’s accounting principles, the reasonableness of significant judgments and the clarity of the disclosures in the Financial Statements. |
|
(g) |
Review any news releases and reports to be issued by the Corporation containing earnings guidance or financial information for research, analysts and rating agencies. The Audit Committee shall also review the Corporation’s policies relating to financial disclosure and the release of earnings guidance and the Corporation’s compliance with financial disclosure rules and regulations. |
|
(h) |
Review any errors or omissions in the Financial Statements. |
|
(i) |
Review significant issues affecting financial reports. |
|
(j) |
Review the Corporation’s Annual Report for consistency with the financial disclosure referenced in the annual Financial Statements. |
|
(k) |
Understand how Management develops interim financial information and the nature and extent of external audit involvement. |
|
(l) |
Review the status of material contingent liabilities as reported to the Audit Committee by the Corporation’s Management, and the manner in which any material contingent liability has been disclosed in the Corporation’s Financial Statements. |
|
(m) |
Review any reserves, accruals, provisions, estimates or adopted programs and policies, including factors that affect asset and liability carrying values and the timing of revenue and expense recognition, that may have a material effect upon the Financial Statements. |
|
(n) |
Review the use of special purpose entities and the business purpose and economic effect of off-balance sheet transactions, arrangements, obligations, guarantees and other relationships of the Corporation and their impact on the reported financial results of the Corporation. |
|
(o) |
Review the treatment for financial reporting purposes of any significant transactions which are not a normal part of the Corporation’s operations. |
|
(p) |
Reviewing Management’s determination of tangible or intangible asset impairment, if any, as required by applicable accounting standards. |
|
(q) |
Review emerging developments regarding International Financial Reporting Standards (“IFRS”) (as issued by the IFRS Foundation and the International Accounting Standards Board) that could affect the Corporation. |
|
(r) |
Review the financial reporting obligations of the Corporation pursuant to its by-laws, its borrowing covenants, the Business Corporations Act (British Columbia) and applicable securities regulation and monitor the Corporation’s compliance thereunder. |
|
(s) |
Review with the external auditors the level of co-operation they received from Management, employees and personnel of the Corporation during the audit process, any issues encountered by the auditors and any impediments on the external auditor’s work. |
|
(t) |
Review and resolve any disagreements between Management and the external auditors with respect to accounting practices and principles. |
|
(u) |
Monitor the objectivity and credibility of the Corporation’s financial reports. |
|
2. |
Internal and Disclosure Controls |
|
(a) |
Review and approve corporate signing authorities and modifications thereto. |
|
(b) |
Consider the effectiveness of the Corporation’s internal controls over financial reporting and related information technology security and control. |
|
(c) |
Review with the auditors any issues or concerns related to any internal control systems in the process of the audit. |
|
(d) |
Review the plan and scope of the annual audit with respect to planned reliance and testing of controls and major points contained in the auditor’s management letter resulting from control evaluation and testing. |
|
(e) |
Establish and maintain complaint procedures regarding accounting, internal accounting controls or auditing matters and the confidential anonymous submission by employees of concerns regarding questionable accounting or auditing matters. Such procedures are appended hereto as Appendix A. |
|
(f) |
Review with the Corporation’s Chief Executive Officer and the Chief Financial Officer the Corporation’s disclosure controls and procedures, including any significant deficiencies in, or material non-compliance with, such controls and procedures. |
|
(g) |
Discuss with the Corporation’s Chief Executive Officer and the Chief Financial Officer all elements of certification required pursuant to National Instrument 52-109 – Certification of Disclosure in Issuers’ Annual and Interim Filings. |
|
(h) |
Annually review the Corporation’s Whistleblower Policy and its effectiveness and enforcement. |
|
(i) |
Approve all material related party transactions in advance; of which materiality is set a $1 for such matters. |
|
3. |
Compliance with Legal and Regulatory Requirements |
|
(a) |
Review with Management, external auditors and legal counsel any material litigation claims or other contingencies, including tax assessments, and adequacy of financial provisions, that could materially affect financial reporting. |
|
(b) |
Review with Management and the Board of Directors any issues with regulatory agencies that are likely to have a significant financial impact on the Corporation. |
|
(c) |
Review with counsel the adequacy and effectiveness of the Corporation’s procedures to ensure compliance with the legal and regulatory responsibilities. |
|
(d) |
Review the status of income tax returns and any significant tax issues as they are reported to the Audit Committee by Management or the Board of Directors. |
|
(e) |
Review any inquiries, investigations, or audits of a financial nature by any government, regulatory, or taxation author |
|
4. |
External Audit |
|
(a) |
Oversee the work of the external auditor engaged for the purpose of preparing or issuing an auditor’s report or performing such other audit, review or attest services for the Corporation, including the resolution of disagreements between Management and the external auditor regarding financial reporting. |
|
(b) |
Review and approve the audit plans, scope and proposed audit fees. |
|
(c) |
Discuss with the auditors the results of the audit, any changes in accounting policies or practices and their impact on the financials, as well as any items that might significantly impact financial results. |
|
(d) |
Receive a report from the auditors on critical accounting policies and practices to be used, all alternative treatments of financial information within IFRS that have been discussed with Management, including the ramifications of the use of such alternative treatments, and the treatment preferred by the auditor. |
|
(e) |
Receive an annual report from the auditors describing the audit firm’s internal quality-control procedures, and material issues raised by the most recent internal quality-control review, or peer review, of the firm, or by any inquiry or investigation by governmental or professional authorities, within the preceding five years, respecting one or more audits carried out the firm, and any steps taken to deal with any such issues. |
|
(f) |
Annually review the independence of the external auditors by receiving a report from the independent auditor detailing all relationships between them and the Corporation. In assessing such independence, the Audit Committee shall discuss with the external auditors, and may require a letter from the external auditor outlining any relationships between the external auditors and the Corporation or its affiliates. |
|
(g) |
Review, where there is to be a change of external auditors, all issues related to the change, including the information to be included in the notice of change of auditor called for under National Instrument 51-102 – Continuous Disclosure Obligations or any successor legislation (“NI 51-102”), and the planned steps for an orderly transition. The Audit Committee shall further review all reportable events, including disagreements, unresolved issues and consultations, as defined in NI 51-102 or any successor legislation, on a routine basis, whether or not there is to be a change of external auditor. |
|
(h) |
Separately meet with the auditors, apart from Management, at least once a year. |
|
(i) |
Recommend to the Board of Directors: (i) the external auditor to be nominated for the purpose of preparing or issuing an auditor’s report or performing other audit, review or attest services for the Corporation and, (ii) the compensation of the external auditor. |
|
(j) |
Review, negotiate and recommend to the Board of Directors the execution of all engagement letters of the external auditors, both for audit and non-audit services. |
|
(k) |
Review the performance of the external auditors, including the compensation, scope, and timeliness of the audits and all other related services and any non-audit services provided by the external auditors. |
|
(l) |
Ensure regular rotation of the lead partner and reviewing partner. |
|
(m) |
Establish and oversee policies with regards to the hiring by the Corporation of any partners, employees, and any former partners or employees of any present or former firms that acted as external auditors of the Corporation. |
|
5. |
Non-Audit Services |
|
(a) |
Pre-approve all non-audit services to be provided to the Corporation or its subsidiary entities by the external auditor. Pre-approval may be granted by any one Member of the Audit Committee. |
|
6. |
Risk Management |
|
(a) |
Review and monitor the processes in place to identify and manage the principal risks that could impact the financial reporting of the Corporation. |
|
(b) |
Ensure that directors’ and officers’ liability insurance is in place. |
|
(c) |
Review and approve corporate investment policies. |
|
(d) |
Assess, as part of its internal controls responsibility, the effectiveness of the over-all process for identifying principal business risks and report thereon to the Board of Directors. |
|
7. |
Other Responsibilities and Matters |
|
(a) |
Ensure that this Charter or an appropriate summary of it which has been granted approval by the Audit Committee is properly disclosed in accordance with any securities laws or regulatory requirements. |
|
(b) |
Review annually the adequacy of this Charter and confirm that all responsibilities have been carried out. |
|
(c) |
Evaluate the Audit Committee’s and individual Member’s performance on a regular basis and report annually to the Board of Directors the result of its annual self-assessment. |
|
(d) |
Review and approve the Corporation’s hiring policies regarding partners, employees and former partners and employees of the present and former external auditor of the Corporation. |
|
(e) |
Review the appointments of the Corporation’s Chief Financial Officer, internal auditor (or persons appointed to perform the internal audit function), and any key financial executives involved in the financial reporting process of the Corporation and any material subsidiary. |
|
(f) |
Discuss the Corporation’s compliance with tax and financial reporting laws and regulation, if and when issues arise. |
|
(g) |
Review all material balance sheet issues, material contingent obligations and material related party transactions. |
|
(h) |
Periodically assess the Corporation’s need for an internal audit function, if not present. |
|
(i) |
Take such other actions within the general scope of its responsibilities as the Audit Committee shall deem appropriate or as directed by the Board of Directors. |
Authority
The Audit Committee shall have full access to all of the Corporation’s books, records, properties, facilities and personnel, subject to compliance with any leases or similar contacts governing same.
Additionally, the Audit Committee has the authority to engage independent counsel and other advisors as it determines necessary to carry out its duties and to set and pay the compensation for any advisors employed by the Audit Committee at the cost of the Corporation without obtaining approval of the Board of Directors, based on its sole judgment and discretion. The Audit Committee has the authority to communicate directly with the internal and external auditors of the Corporation.
Inconsistencies with Applicable Laws
In the event of any conflict or inconsistency between this Charter and the applicable laws, in each case as amended, restated or amended and restated from time to time, the provisions hereof shall be ineffective and shall be superseded by the provisions of such applicable laws to the extent necessary to resolve such conflict or inconsistency.
Appendix A
To Audit Committee Charter
Procedures for the Submission of Complaints or Concerns
Regarding Accounting, Internal Accounting Controls or Auditing Matters
|
1. |
The Corporation shall forward to the Audit Committee of the Board of Directors any complaints that it has received regarding accounting, internal accounting controls or auditing matters. |
|
2. |
Any employee of the Corporation may submit, on a confidential, anonymous basis if the employee so desires, any concerns by sending such concerns in writing and forwarding them in a sealed envelope to: |
Attention: Chair of the Audit Committee
Bright Minds Biosciences Inc.
1500 – 1055 West Georgia Street
Vancouver, BC
V6E 4N7
The envelope is to be clearly marked: “To be opened by the Audit Committee only.”
Any such envelopes shall be forwarded promptly to the Chair.
|
3. |
Contact information including a phone number and e-mail address shall be published for the Chair on the Corporation’s website for those people wishing to contact the Chair directly. |
|
4. |
At each of its meetings following the receipt of any information pursuant to this Appendix, the Audit Committee shall review and consider any such complaints or concerns and take any action that it deems appropriate in the circumstances. |
|
5. |
The Audit Committee shall retain any such complaints or concerns along with the material gathered to support its actions for a period of no less than seven years. Such records will be held on behalf of the Audit Committee by the Audit Committee Secretary. |
|
6. |
This Appendix A shall appear on the Corporation’s website as part of this Charter. |