
.2
Targeted therapies for people of all ages January 2026 Day One Biopharmaceuticals

2 Disclaimer This presentation and the accompanying oral commentary contain forward - looking statements that are based on our management’s bel iefs and assumptions and on information currently available to our management. Forward - looking statements are inherently subject to risks and uncertainties, some of whi ch cannot be predicted or quantified. In some cases, you can identify forward - looking statements by terminology such as “may,” “will,” “should,” “could,” “expect,” “plan,” anticipate,” “believe,” “estimate,” “predict,” “intend,” “potential,” “would,” “continue,” “ongoing” or the negative of these terms or other comparab le terminology. Forward - looking statements include all statements other than statements of historical fact contained in this presentation, including information concerning our fut ure financial performance, including the sufficiency of our cash, cash equivalents and short - term investments to fund our operations, business plans and objectives, timi ng and success of our commercialization and marketing efforts, timing and success of our planned nonclinical and clinical development activities, the success of our acquisition of Mersana Therapeutics and its Emi - Le program, the results of any of our strategic collaborations, including the potential achievement of milestones and provision of royalty payments thereunder, efficacy and safety profiles of our products and product candidates, the ability of OJEMDA ( tovorafenib ) to treat pediatric low - grade glioma (pLGG) or related indications, the potential therapeutic benefits and economic value of our products and product candidates, potential growth opportunities, competitive position, ind ust ry environment and potential market opportunities, our ability to protect intellectual property and the impact of global business or macroeconomic conditions, in clu ding as a result of inflation, changing interest rates, government shutdowns, cybersecurity incidents, significant political, trade or regulatory developments, including tari ffs , shifting priorities within the U.S. Food and Drug Administration and reduced funding of federal healthcare programs, and global regional conflicts, on our business and operati ons . Forward - looking statements are subject to known and unknown risks, uncertainties, assumptions and other factors. It is not possi ble for our management to predict all risks, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may ca use actual results to differ materially from those contained in any forward - looking statements we may make. These factors, together with those that are described under the heading “Risk Factors” contained in our most recent Quarterly Report on Form 10 - Q filed with the Securities and Exchange Commission (SEC) and other documents we file from time to t ime with the SEC, may cause our actual results, performance or achievements to differ materially and adversely from those anticipated or implied by our forward - looking statements. In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. Th ese statements are based upon information available to us as of the date of this presentation, and although we believe such information forms a reasonable basis for su ch statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted a thorough inquiry into, or review of, a ll potentially available relevant information. These statements are inherently uncertain and investors are cautioned not to unduly rely upon these statements. Furthermore, if our fo rward - looking statements prove to be inaccurate, the inaccuracy may be material. In light of the significant uncertainties in these forward - looking statements, you s hould not regard these statements as a representation or warranty by us or any other person that we will achieve our objectives and plans in any specified time fram e, or at all. We undertake no obligation to publicly update any forward - looking statements, whether as a result of new information, future events or otherwise, except as required by law. This presentation also contains estimates and other statistical data made by independent parties and by us relating to market si ze and growth and other data about our industry. This data involves a number of assumptions and limitations, and you are cautioned not to give undue weight to such est imates. In addition, projections, assumptions and estimates of our future performance and the future performance of the markets in which we operate are necessarily subject to a high degree of uncertainty and risk.

3 Inspired by the urgent needs of children, Day One creatively and intentionally develops new medicines for people of all ages with life - threatening diseases

Bringing life - changing medicines to patients sooner 4 • Commercial - stage biopharmaceutical company • Our goal is to develop and provide access to targeted new medicines to patients of all ages as rapidly as possible • Focused on advancing first - or best - in - class medicines for childhood and adult diseases Who we are OJEMDA received approval in April 2024 and is indicated for the treatment of pediatric patients 6 months of age and older wit h r elapsed or refractory pediatric low - grade glioma harboring a BRAF fusion or rearrangement, or BRAF V600 mutation. 2021 IPO 2018 FOUNDED 2024 OJEMDA TM APPROVAL Nasdaq: DAWN
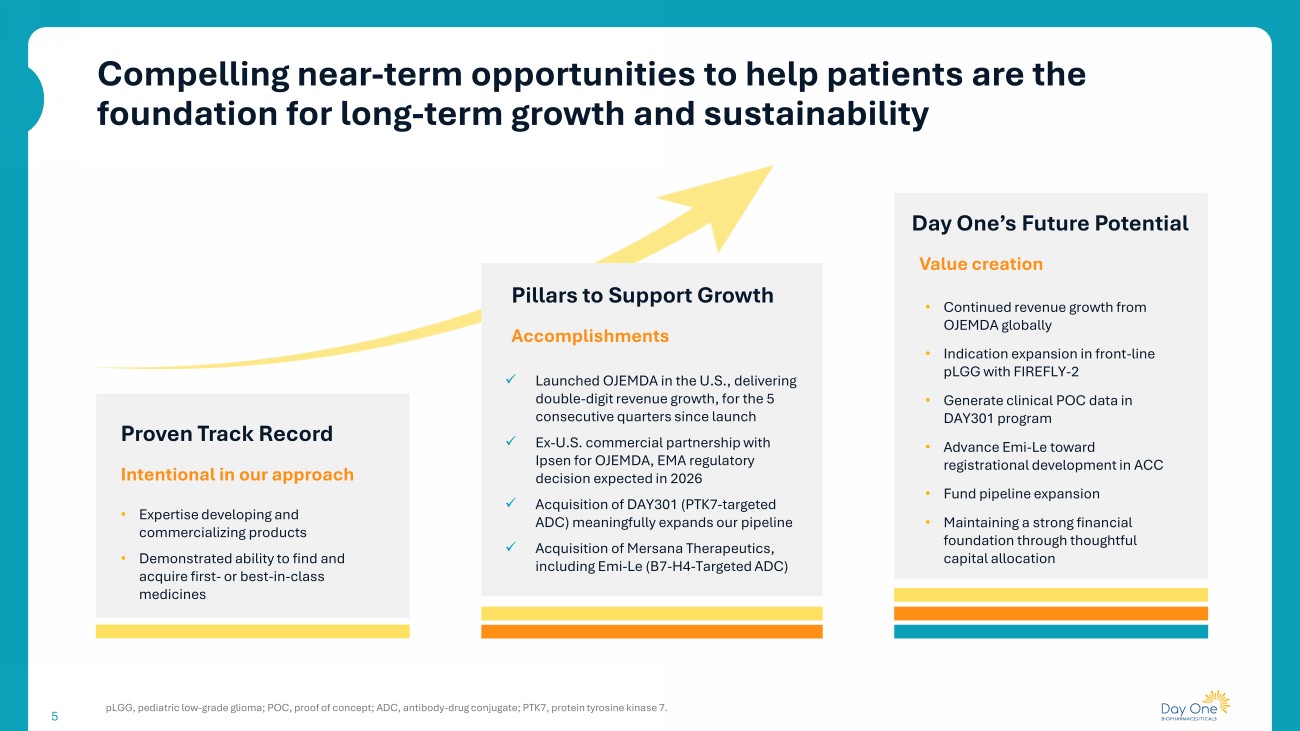
5 Day One’s Future Potential Value creation • Continued revenue growth from OJEMDA globally • Indication expansion in front - line pLGG with FIREFLY - 2 • Generate clinical POC data in DAY301 program • Advance Emi - Le toward registrational development in ACC • Fund pipeline expansion • Maintaining a strong financial foundation through thoughtful capital allocation Proven Track Record Intentional in our approach • Expertise developing and commercializing products • Demonstrated ability to find and acquire first - or best - in - class medicines Pillars to Support Growth Accomplishments x Launched OJEMDA in the U.S., delivering double - digit revenue growth, for the 5 consecutive quarters since launch x Ex - U.S. commercial partnership with Ipsen for OJEMDA, EMA regulatory decision expected in 2026 x Acquisition of DAY301 (PTK7 - targeted ADC) meaningfully expands our pipeline x Acquisition of Mersana Therapeutics, including Emi - Le (B7 - H4 - Targeted ADC) Compelling near - term opportunities to help patients are the foundation for long - term growth and sustainability pLGG, pediatric low - grade glioma; POC, proof of concept; ADC, antibody - drug conjugate; PTK7, protein tyrosine kinase 7. 5
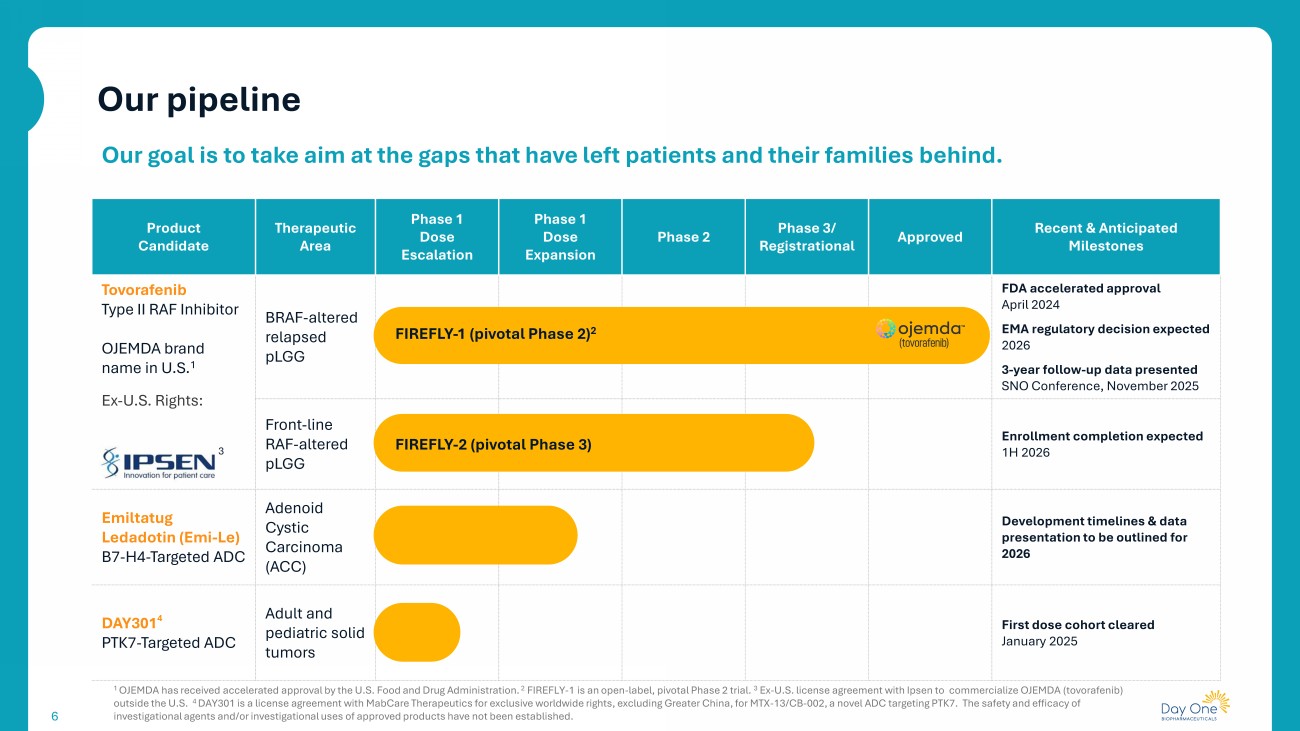
Recent & Anticipated Milestones Approved Phase 3/ Registrational Phase 2 Phase 1 Dose Expansion Phase 1 Dose Escalation Therapeutic Area Product Candidate FDA accelerated approval April 2024 EMA regulatory decision expected 2026 3 - year follow - up data presented SNO Conference, November 2025 BRAF - altered relapsed pLGG Tovorafenib Type II RAF Inhibitor OJEMDA brand name in U.S. 1 Ex - U.S. Rights: 3 Enrollment completion expected 1H 2026 Front - line RAF - altered pLGG Development timelines & data presentation to be outlined for 2026 Adenoid Cystic Carcinoma (ACC) Emiltatug Ledadotin (Emi - Le) B7 - H4 - Targeted ADC First dose cohort cleared January 2025 Adult and pediatric solid tumors DAY301 4 PTK7 - Targeted ADC 6 Our goal is to take aim at the gaps that have left patients and their families behind. FIREFLY - 1 (pivotal Phase 2) 2 FIREFLY - 2 (pivotal Phase 3) 1 OJEMDA has received accelerated approval by the U.S. Food and Drug Administration. 2 FIREFLY - 1 is an open - label, pivotal Phase 2 trial. 3 Ex - U.S. license agreement with Ipsen to commercialize OJEMDA ( tovorafenib ) outside the U.S. 4 DAY301 is a license agreement with MabCare Therapeutics for exclusive worldwide rights, excluding Greater China, for MTX - 13/CB - 002, a novel ADC targeting PTK7. The safet y and efficacy of investigational agents and/or investigational uses of approved products have not been established. Our pipeline

Relapsed or refractory BRAF - altered pLGG OJEMDA 7 Nora Living with pLGG

8 A serious and life - threatening disease *Incidence of BRAF alterations varies across pLGG subtypes. 1 Sievert AJ, Fisher MJ. Pediatric low - grade gliomas. J Child Neurol. 2009;24(11): 1397 - 1408 . doi:10.1177/0883073809342005 . 2 Penman CL et al. Front Oncol . 2015;5:54. 3 Cohen AR ., N Engl J Med. 2020;386(20):1922 - 1931. 4 Lassaletta A, et al. J Clin Oncol . 2017;35(25):2934 - 2941. 5 Faulkner C, et al. J Neuropathol Exp Neurol. 2015;74(9):867 - 872. 6 Packer RJ, et al. Neuro Oncol. 2017;19(6):750 - 761. 7 Ostrum QT et al., Neuro Oncol. 2015; 16( Suppl 10):x1 - x36; 8 De Blank P. et al., Curr Opin Pediatr . 2019 Feb; 31(1):21 - 27. Pediatric low - grade glioma: The most common type of brain tumor in children • For the majority of patients with pLGG in the relapsed setting, there is no standard of care, and until recently, no approved therapies • Up to 75% of pLGGs have a BRAF alteration*, of those ~80% are BRAF fusions and ~20% are BRAF V600 mutations 2 - 6 • Despite surgery playing a significant role in treatment, the vast majority of patients still require systemic therapy 7,8 • Due to high rate of disease recurrence, most patients will undergo multiple lines of systemic therapy over the course of their disease pLGGs are chronic and relentless , with patients suffering profound tumor and treatment - associated morbidity that can impact their life trajectory over the long term 1

9 Available in tablet formulation and pediatric - friendly powder for oral suspension Overview U.S. prescribing information for OJEMDA 9 Indication OJEMDA is indicated for the treatment of pediatric patients 6 months of age and older with relapsed or refractory pediatric low - grade glioma harboring a BRAF fusion or rearrangement, or BRAF V600 mutation Recommended Dose 380 mg/m 2 administered orally once weekly (not to exceed a dose of 600mg once weekly); OJEMDA can be taken with or without food For full prescribing information, visit dayonebio.com * This indication is approved under accelerated approval based on response rate and duration of response. Continued approval fo r t his indication may be contingent upon verification of clinical benefit in a confirmatory trial.

10 Data from Pivotal Phase 2 FIREFLY - 1 trial. • Meaningful tumor stabilization or shrinkage may be possible with OJEMDA, in the clinical trial: • 51% of children experienced tumor shrinkage by at least 25% • 82% of children saw their tumors shrink or remain stable Efficacy Safety • Generally well - tolerated therapy, with 9 out of 10 patients staying on treatment in the clinical trial • Most common grade 3 / 4 adverse events include: anemia, elevated CPK, maculo - papular rash, fatigue & vomiting Dosing • Once - weekly, taken with or without food conveniently from home can mean fewer daily interruptions OJEMDA is indicated for the treatment of patients 6 months of age and older with relapsed or refractory pediatric low - grade glio ma (pLGG) harboring a BRAF fusion, rearrangement, or BRAF V600 mutation. Product profile aligns with what physicians are looking for in a therapy
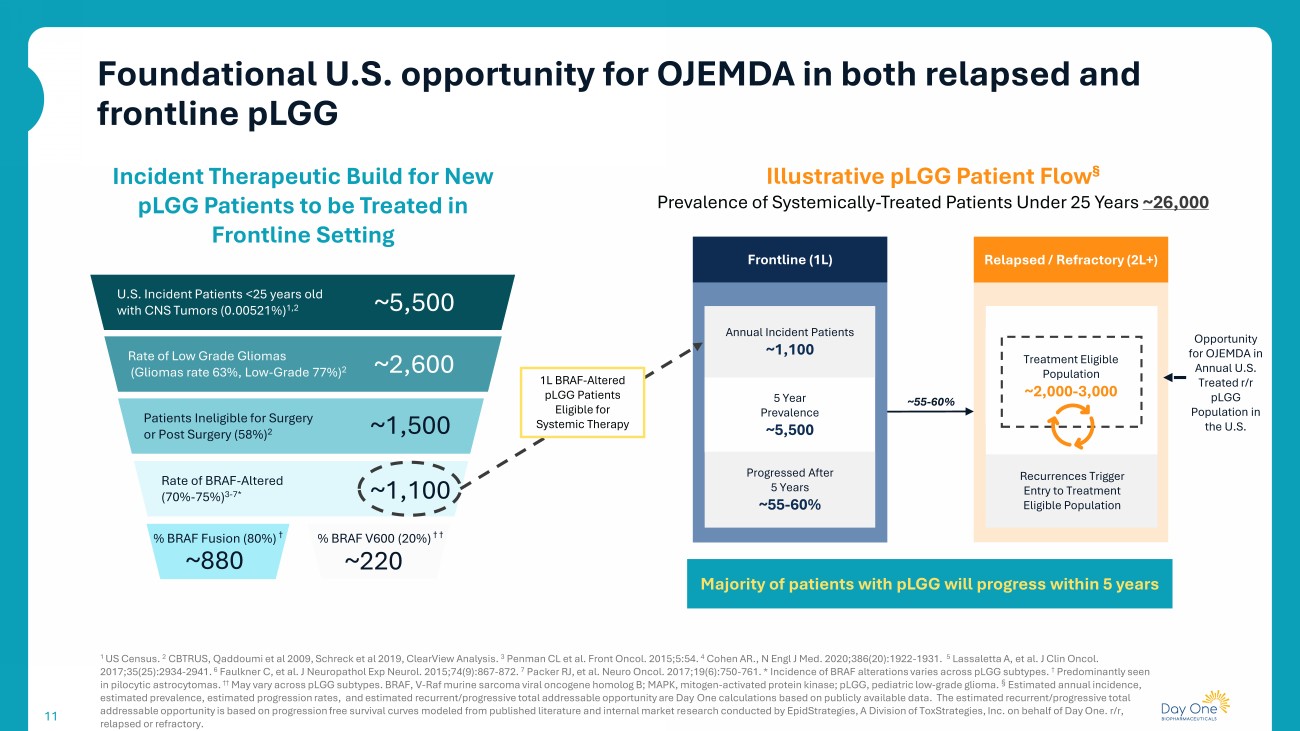
11 1 US Census. 2 CBTRUS, Qaddoumi et al 2009, Schreck et al 2019, ClearView Analysis. 3 Penman CL et al. Front Oncol. 2015;5:54. 4 Cohen AR., N Engl J Med. 2020;386(20):1922 - 1931. 5 Lassaletta A, et al. J Clin Oncol. 2017;35(25):2934 - 2941. 6 Faulkner C, et al. J Neuropathol Exp Neurol. 2015;74(9):867 - 872. 7 Packer RJ, et al. Neuro Oncol. 2017;19(6):750 - 761. * Incidence of BRAF alterations varies across pLGG subtypes. † Predominantly seen in pilocytic astrocytomas . †† May vary across pLGG subtypes. BRAF, V - Raf murine sarcoma viral oncogene homolog B; MAPK, mitogen - activated protein kinase; pLGG , pediatric low - grade glioma. § Estimated annual incidence, estimated prevalence, estimated progression rates, and estimated recurrent/progressive total addressable opportunity are Day On e calculations based on publicly available data. The estimated recurrent/progressive total addressable opportunity is based on progression free survival curves modeled from published literature and internal market re sea rch conducted by EpidStrategies , A Division of ToxStrategies , Inc. on behalf of Day One. r/r, relapsed or refractory. Incident Therapeutic Build for New pLGG Patients to be Treated in Frontline Setting U.S. Incident Patients <25 years old with CNS Tumors (0.00521%) 1,2 ~5,500 Rate of Low Grade Gliomas (Gliomas rate 63%, Low - Grade 77%) 2 ~2,600 ~1,500 Patients Ineligible for Surgery or Post Surgery (58%) 2 ~1,100 % BRAF Fusion (80%) † % BRAF V600 (20%) † † ~880 ~220 Frontline (1L) Annual Incident Patients ~1,100 1L BRAF - Altered pLGG Patients Eligible for Systemic Therapy Illustrative pLGG Patient Flow § Prevalence of Systemically - Treated Patients Under 25 Years ~26,000 5 Year Prevalence ~5,500 Progressed After 5 Years ~55 - 60% Relapsed / Refractory (2L+) ~55 - 60% Majority of patients with pLGG will progress within 5 years Opportunity for OJEMDA in Annual U.S. Treated r/r pLGG Population in the U.S. Rate of BRAF - Altered (70% - 75%) 3 - 7* Treatment Eligible Population ~2,000 - 3,000 Recurrences Trigger Entry to Treatment Eligible Population Foundational U.S. opportunity for OJEMDA in both relapsed and frontline pLGG 11
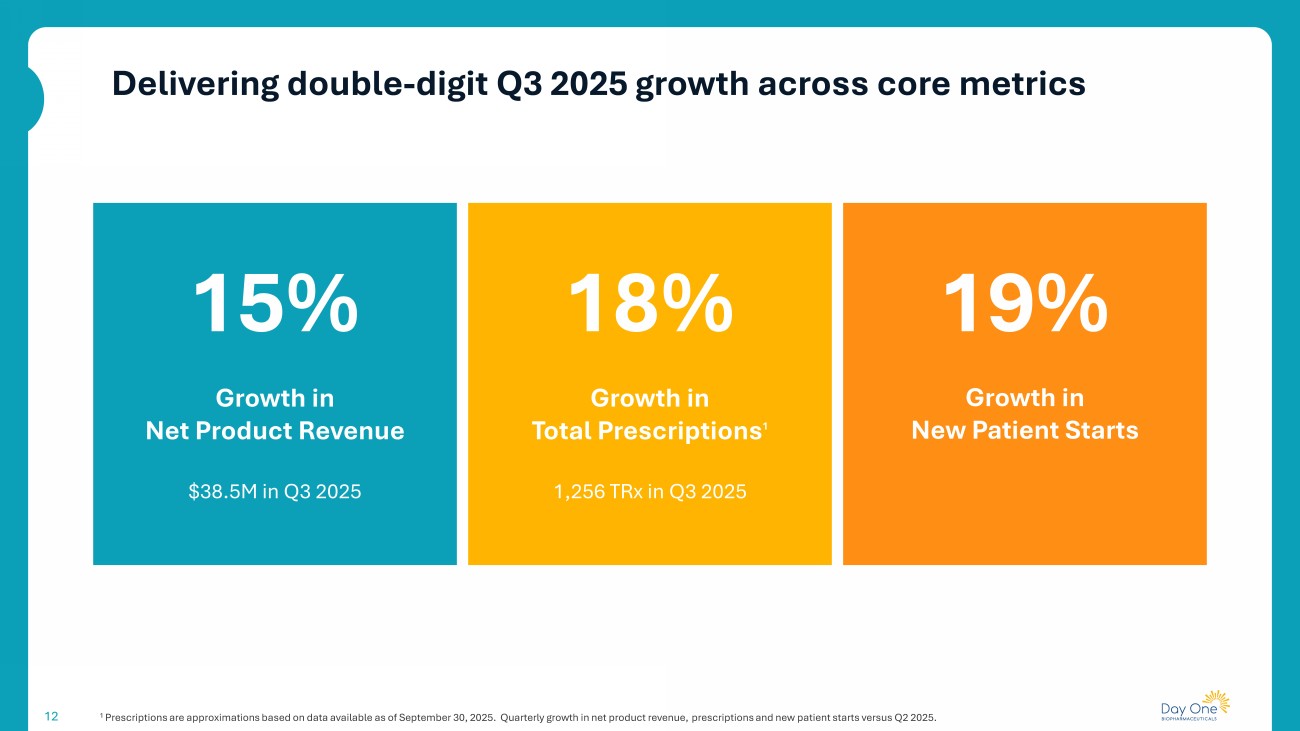
Delivering double - digit Q3 2025 growth across core metrics 12 1 Prescriptions are approximations based on data available as of September 30, 2025. Quarterly growth in net product revenue, pre scriptions and new patient starts versus Q2 2025. 15% Growth in Net Product Revenue 18% $38.5M in Q3 2025 19% Growth in New Patient Starts Growth in Total Prescriptions 1 1,256 TRx in Q3 2025
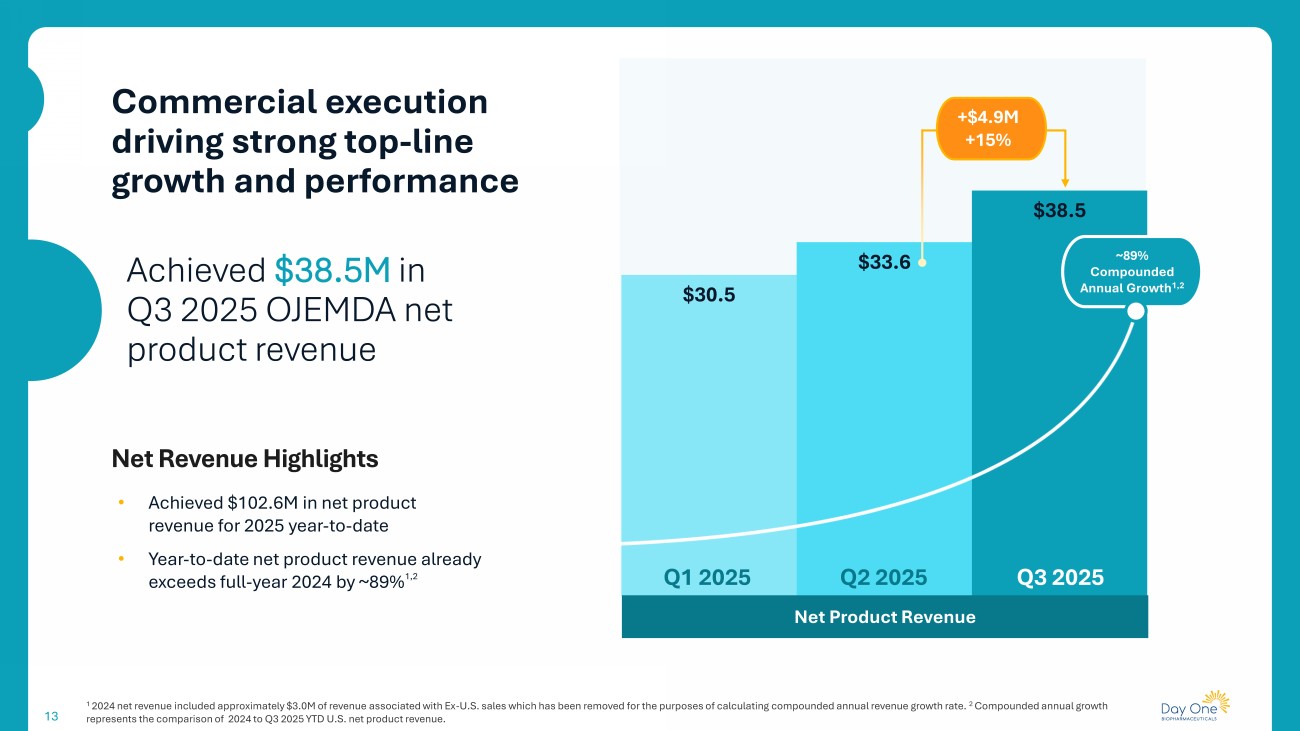
13 1 2024 net revenue included approximately $3.0M of revenue associated with Ex - U.S. sales which has been removed for the purposes o f calculating compounded annual revenue growth rate. 2 Compounded annual growth represents the comparison of 2024 to Q3 2025 YTD U.S. net product revenue. Commercial execution driving strong top - line growth and performance $30.5 $33.6 $38.5 Net Product Revenue Q3 2025 Q1 2025 Q2 2025 +$4.9M +15% ~89% Compounded Annual Growth 1,2 Achieved $38.5M in Q3 2025 OJEMDA net product revenue Net Revenue Highlights • Achieved $102.6M in net product revenue for 2025 year - to - date • Year - to - date net product revenue already exceeds full - year 2024 by ~89% 1,2
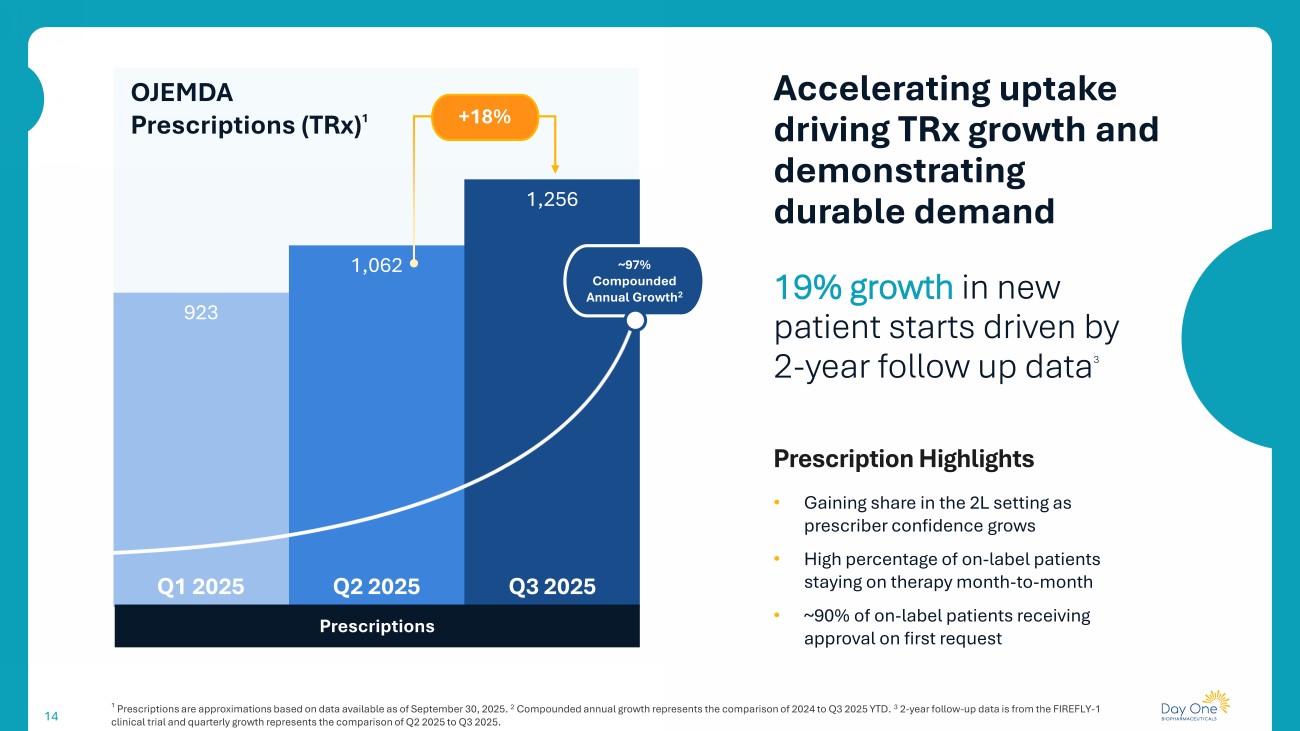
923 1,062 1,256 14 19% growth in new patient starts driven by 2 - year follow up data 3 Prescription Highlights • Gaining share in the 2L setting as prescriber confidence grows • High percentage of on - label patients staying on therapy month - to - month • ~90% of on - label patients receiving approval on first request Accelerating uptake driving TRx growth and demonstrating durable demand Prescriptions OJEMDA Prescriptions ( TRx ) 1 Q3 2025 Q1 2025 Q2 2025 ¹ Prescriptions are approximations based on data available as of September 30, 2025. 2 Compounded annual growth represents the comparison of 2024 to Q3 2025 YTD. 3 2 - year follow - up data is from the FIREFLY - 1 clinical trial and quarterly growth represents the comparison of Q2 2025 to Q3 2025. ~97% Compounded Annual Growth 2 +18%
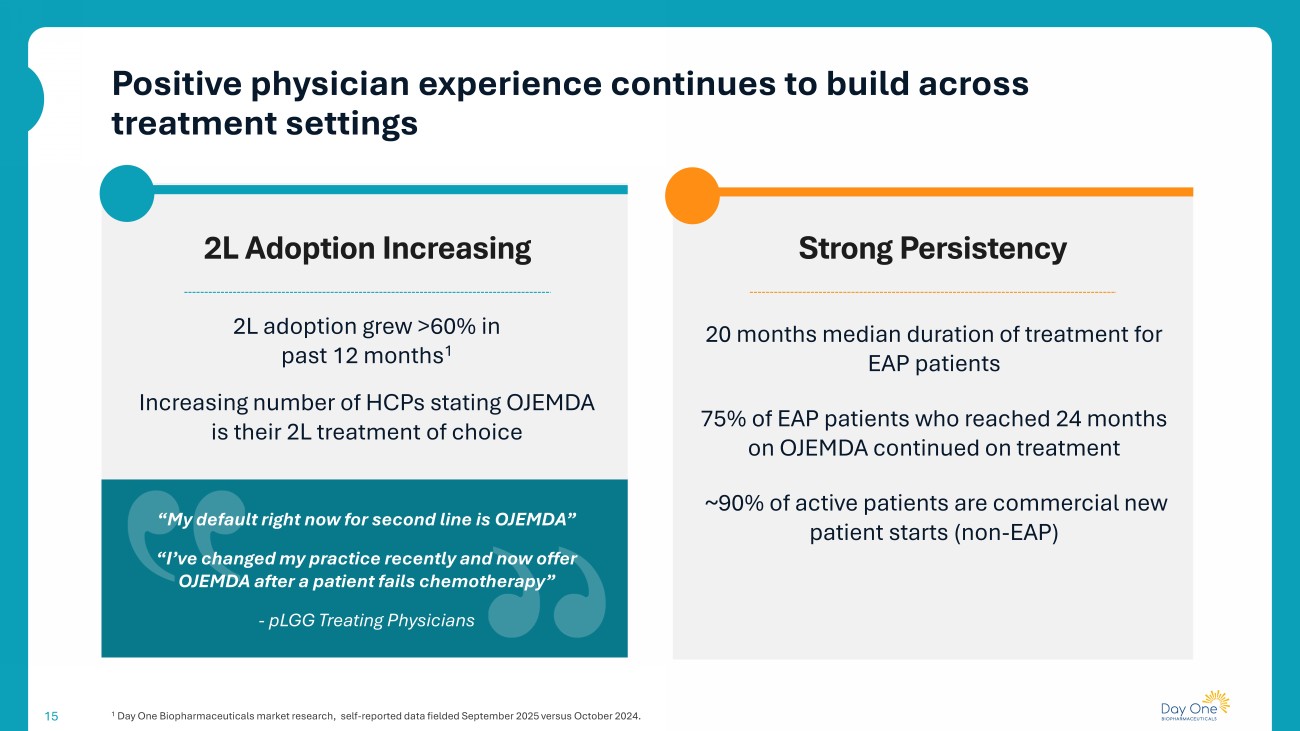
Positive physician experience continues to build across treatment settings 15 1 Day One Biopharmaceuticals market research, self - reported data fielded September 2025 versus October 2024. Strong Persistency 2L Adoption Increasing 20 months median duration of treatment for EAP patients 75% of EAP patients who reached 24 months on OJEMDA continued on treatment ~90% of active patients are commercial new patient starts (non - EAP) 2L adoption grew >60% in past 12 months 1 Increasing number of HCPs stating OJEMDA is their 2L treatment of choice “My default right now for second line is OJEMDA” “I’ve changed my practice recently and now offer OJEMDA after a patient fails chemotherapy” - pLGG Treating Physicians
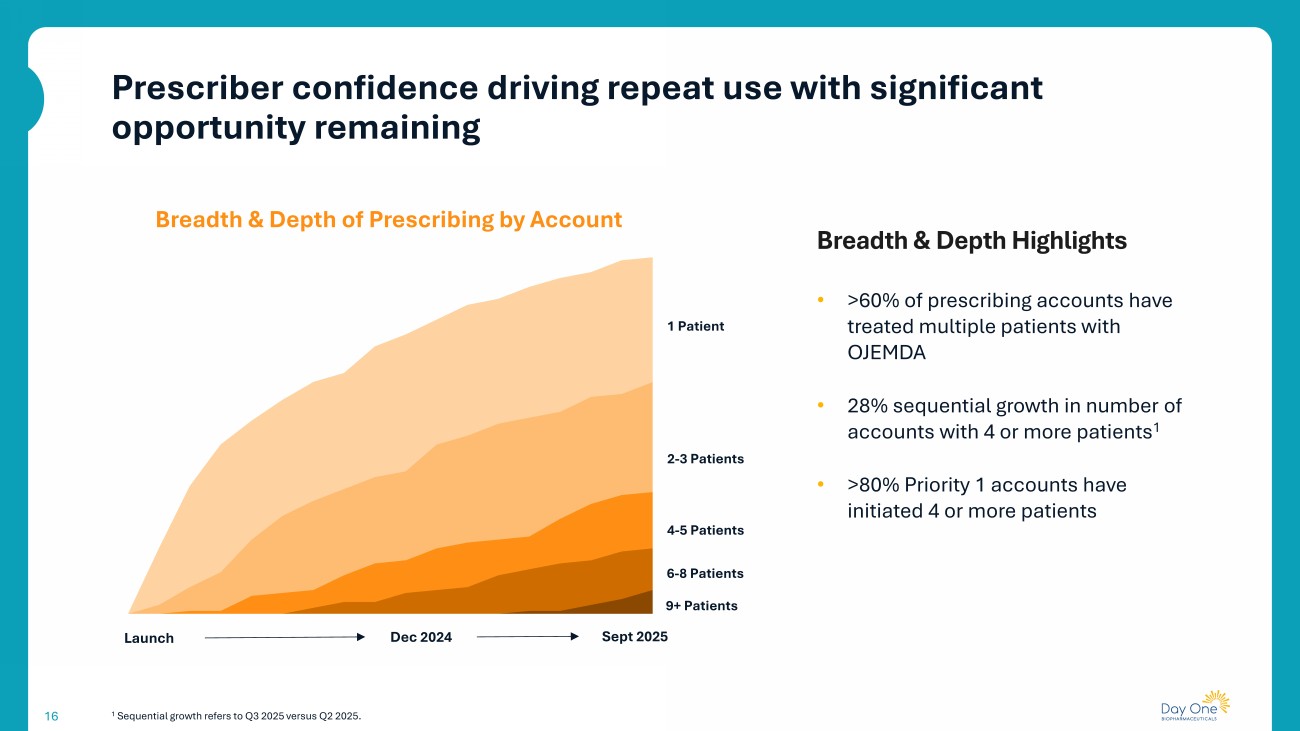
16 1 Sequential growth refers to Q3 2025 versus Q2 2025. • >60% of prescribing accounts have treated multiple patients with OJEMDA • 28% sequential growth in number of accounts with 4 or more patients 1 • >80% Priority 1 accounts have initiated 4 or more patients Launch Dec 2024 Sept 2025 1 Patient 2 - 3 Patients 4 - 5 Patients 6 - 8 Patients 9+ Patients Breadth & Depth of Prescribing by Account Breadth & Depth Highlights Prescriber confidence driving repeat use with significant opportunity remaining
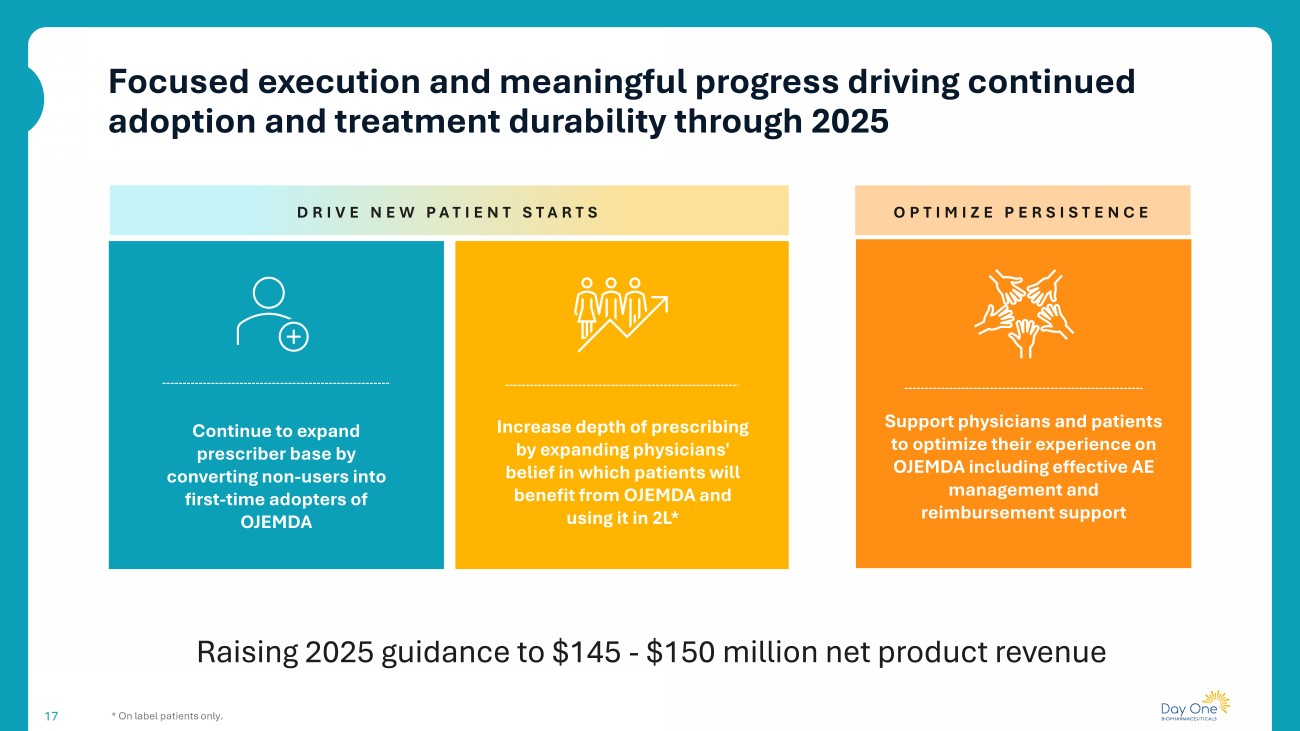
Focused execution and meaningful progress driving continued adoption and treatment durability through 2025 17 * On label patients only. Raising 2025 guidance to $145 - $150 million net product revenue OPTIMIZE PERSISTENCE Support physicians and patients to optimize their experience on OJEMDA including effective AE management and reimbursement support Increase depth of prescribing by expanding physicians' belief in which patients will benefit from OJEMDA and using it in 2L* Continue to expand prescriber base by converting non - users into first - time adopters of OJEMDA DRIVE NEW PATIENT STARTS

We continue to strengthen the OJEMDA story through enhancing the target product profile and data generation 18 FIREFLY - 1 3 - Year Data Highlighting clinical stability following treatment with OJEMDA and opportunity for retreatment Potential to further expand physician confidence and adoption while reinforcing target product profile Presentation at SNO conference in November 2025; publication efforts underway Establishing OJEMDA as the Standard of Care in 2L r/r BRAF - altered pLGG
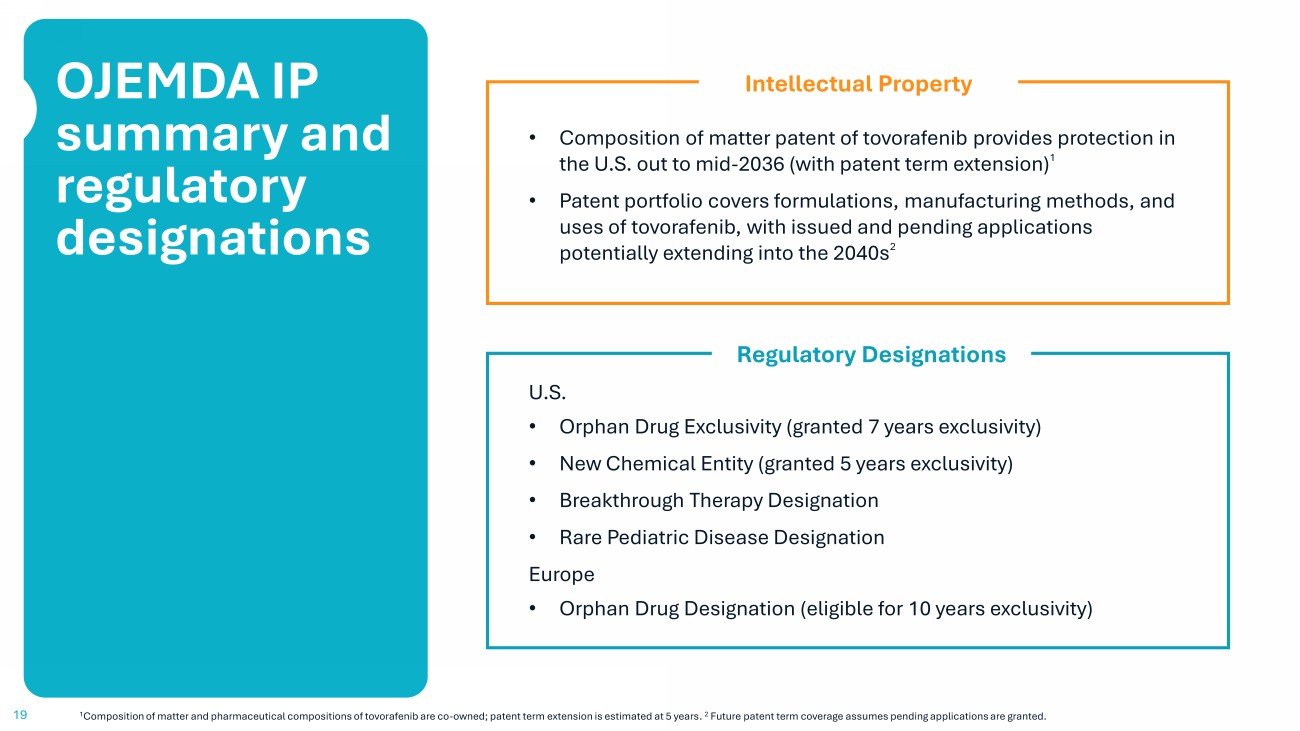
19 OJEMDA IP summary and regulatory designations • Composition of matter patent of tovorafenib provides protection in the U.S. out to mid - 2036 (with patent term extension) 1 • Patent portfolio covers formulations, manufacturing methods, and uses of tovorafenib, with issued and pending applications potentially extending into the 2040s 2 1 Composition of matter and pharmaceutical compositions of tovorafenib are co - owned; patent term extension is estimated at 5 years . 2 Future patent term coverage assumes pending applications are granted. Intellectual Property U.S. • Orphan Drug Exclusivity (granted 7 years exclusivity) • New Chemical Entity (granted 5 years exclusivity) • Breakthrough Therapy Designation • Rare Pediatric Disease Designation Europe • Orphan Drug Designation (eligible for 10 years exclusivity) Regulatory Designations

Phase 2 FIREFLY - 1 Trial 20 3 - year follow - up data from the FIREFLY - 1 trial studying OJEMDA
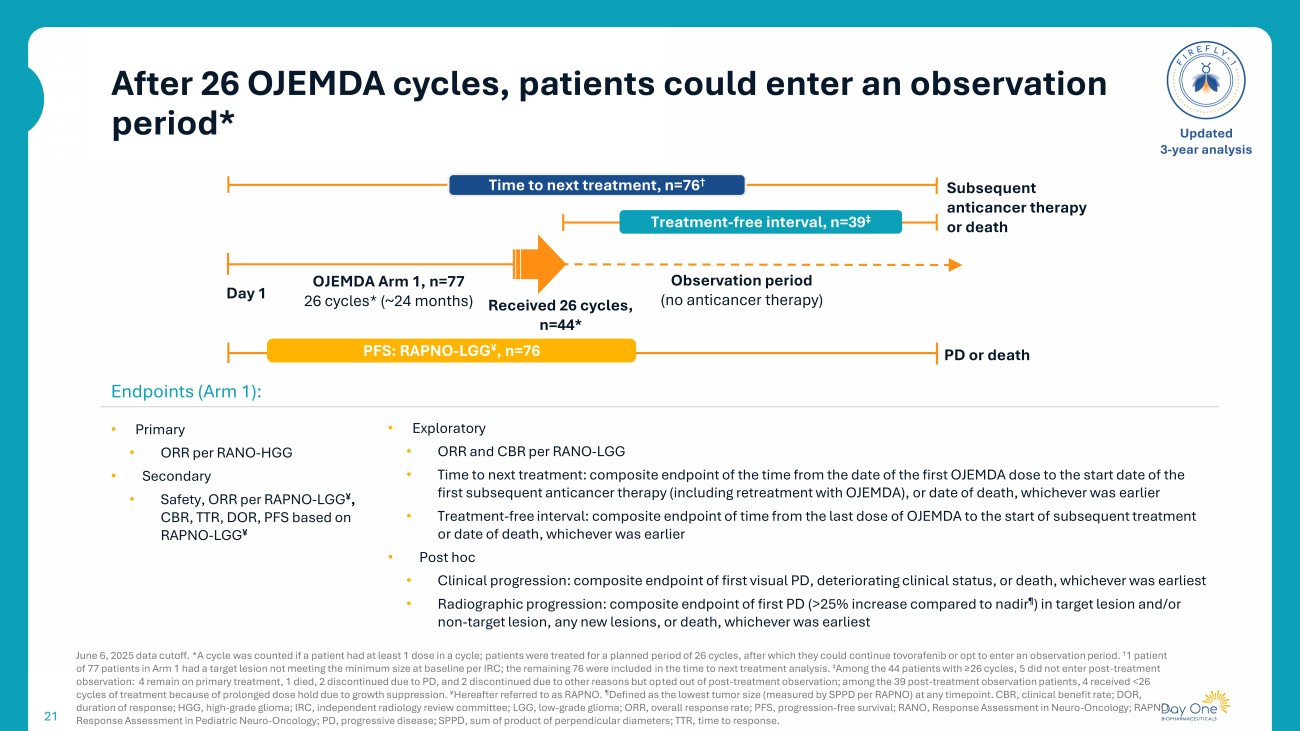
After 26 OJEMDA cycles, patients could enter an observation period* 21 June 6, 2025 data cutoff. *A cycle was counted if a patient had at least 1 dose in a cycle; patients were treated for a planned period of 26 cycles, after which they could continue tovorafenib or opt to enter an observation period. † 1 patient of 77 patients in Arm 1 had a target lesion not meeting the minimum size at baseline per IRC; the remaining 76 were included in the time to next treatment analysis. ‡ Among the 44 patients with ≥26 cycles, 5 did not enter post - treatment observation: 4 remain on primary treatment, 1 died, 2 discontinued due to PD, and 2 discontinued due to other reasons but op ted out of post - treatment observation; among the 39 post - treatment observation patients, 4 received <26 cycles of treatment because of prolonged dose hold due to growth suppression. ¥ Hereafter referred to as RAPNO. ¶ Defined as the lowest tumor size (measured by SPPD per RAPNO) at any timepoint. CBR, clinical benefit rate; DOR, duration of response; HGG, high - grade glioma; IRC, independent radiology review committee; LGG, low - grade glioma; ORR, overall r esponse rate; PFS, progression - free survival; RANO, Response Assessment in Neuro - Oncology ; RAPNO, Response Assessment in Pediatric Neuro - Oncology; PD, progressive disease; SPPD, sum of product of perpendicular diameters; TTR, time to response . Updated 3 - year analysis Endpoints (Arm 1): • Primary • ORR per RANO - HGG • Secondary • Safety, ORR per RAPNO - LGG ¥ , CBR, TTR, DOR, PFS based on RAPNO - LGG ¥ • Exploratory • ORR and CBR per RANO - LGG • Time to next treatment: composite endpoint of the time from the date of the first OJEMDA dose to the start date of the first subsequent anticancer therapy (including retreatment with OJEMDA), or date of death, whichever was earlier • Treatment - free interval: composite endpoint of time from the last dose of OJEMDA to the start of subsequent treatment or date of death, whichever was earlier • Post hoc • Clinical progression: composite endpoint of first visual PD, deteriorating clinical status, or death, whichever was earliest • Radiographic progression: composite endpoint of first PD (>25% increase compared to nadir ¶ ) in target lesion and/or non - target lesion, any new lesions, or death, whichever was earliest Subsequent anticancer therapy or death OJEMDA Arm 1, n=77 26 cycles* (~24 months) Received 26 cycles, n=44* Observation period ( no anticancer therapy) Time to next treatment, n=76 † Treatment - free inter val , n=39 ‡ Day 1 PD or death PFS: RAPNO - LGG ¥ , n=76
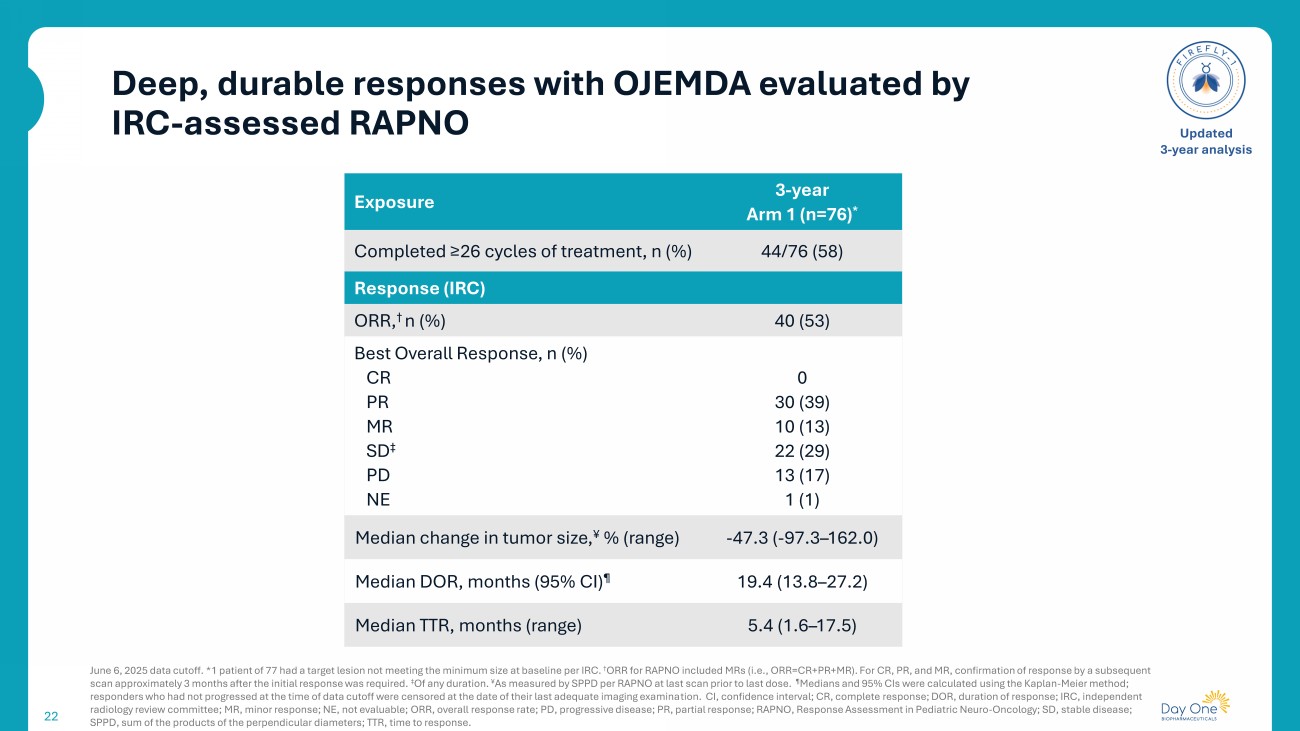
Deep, durable responses with OJEMDA evaluated by IRC - assessed RAPNO 22 June 6, 2025 data cutoff. * 1 patient of 77 had a target lesion not meeting the minimum size at baseline per IRC. † ORR for RAPNO included MRs (i.e., ORR=CR+PR+MR). For CR, PR, and MR, confirmation of response by a subsequent scan approximately 3 months after the initial response was required. ‡ Of any duration. ¥ As measured by SPPD per RAPNO at last scan prior to last dose. ¶ Medians and 95% CIs were calculated using the Kaplan - Meier method; responders who had not progressed at the time of data cutoff were censored at the date of their last adequate imaging examina tio n. CI, confidence interval; CR, complete response; DOR, duration of response; IRC, independent radiology review committee ; MR, minor response; NE, not evaluable; ORR, overall response rate; PD, progressive disease; PR, partial response; RAPNO, Response Assessment in Pediatric Neuro - Oncology ; SD, stable disease; SPPD, sum of the products of the perpendicular diameters; TTR, time to response. Updated 3 - year analysis 3 - year Arm 1 ( n=76) * Exposure 44/76 (58) Completed ≥26 cycles of treatment, n (%) Response (IRC) 40 (53) ORR, † n (%) 0 30 (39) 10 (13) 22 (29) 13 (17) 1 (1) Best Overall Response, n (%) CR PR MR SD ‡ PD NE - 47.3 ( - 97.3 – 162.0) Median change in tumor size, ¥ % (range) 19.4 (13.8 – 27.2) Median DOR, months (95% CI) ¶ 5.4 (1.6 – 17.5) Median TTR, months (range)
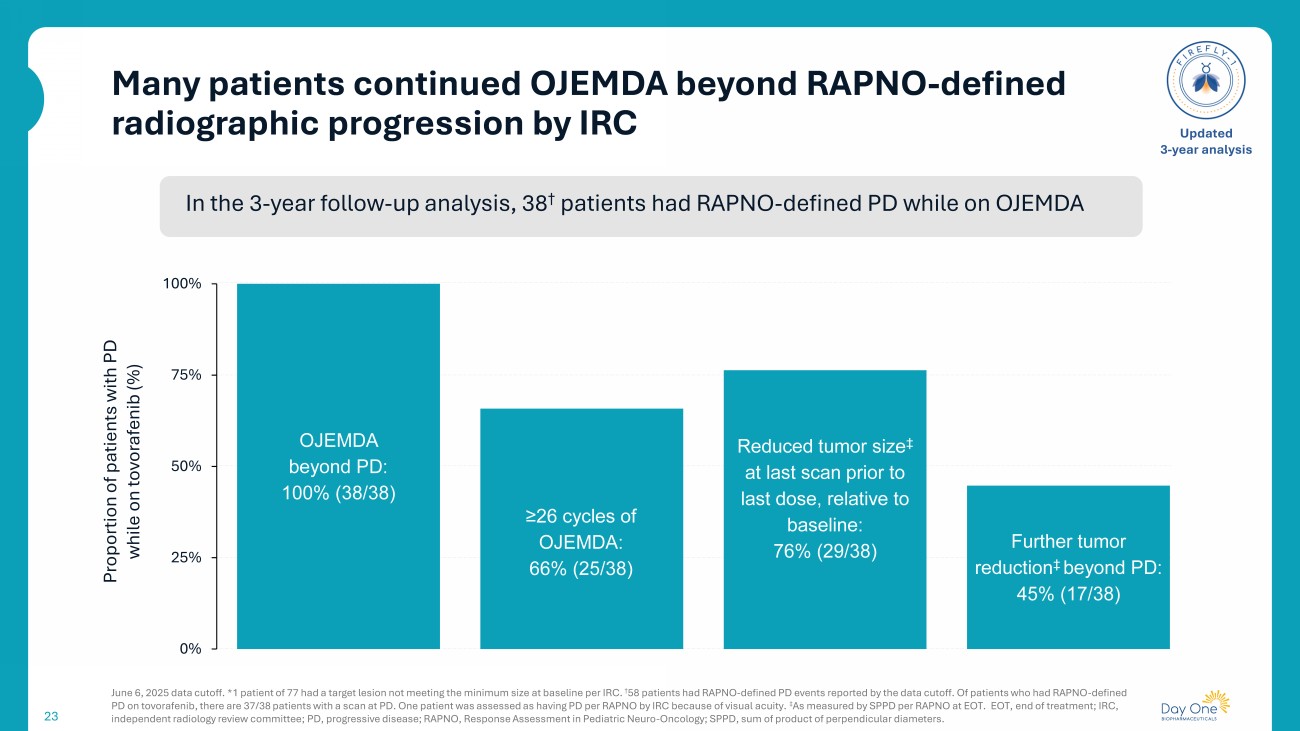
Many patients continued OJEMDA beyond RAPNO - defined radiographic progression by IRC 23 June 6, 2025 data cutoff. * 1 patient of 77 had a target lesion not meeting the minimum size at baseline per IRC. † 58 patients had RAPNO - defined PD events reported by the data cutoff. Of patients who had RAPNO - defined PD on tovorafenib , t here are 37/38 patients with a scan at PD. One patient was assessed as having PD per RAPNO by IRC because of visual acuity. ‡ A s measured by SPPD per RAPNO at EOT. EOT, end of treatment; IRC, independent radiology review committee ; PD, progressive disease; RAPNO, Response Assessment in Pediatric Neuro - Oncology ; SPPD, sum of product of perpendicular diameters. Updated 3 - year analysis OJEMDA beyond PD: 100% ( 38 /38) ≥26 cycles of OJEMDA: 66% ( 25 /38) Reduced tumor size ‡ at last scan prior to last dose, relative to baseline: 76% ( 29 /38) Further tumor reduction ‡ beyond PD: 45% ( 17 /38) 0% 25% 50% 75% 100% Proportion of patients with PD while on tovorafenib (%) In the 3 - year follow - up analysis, 38 † patients had RAPNO - defined PD while on OJEMDA
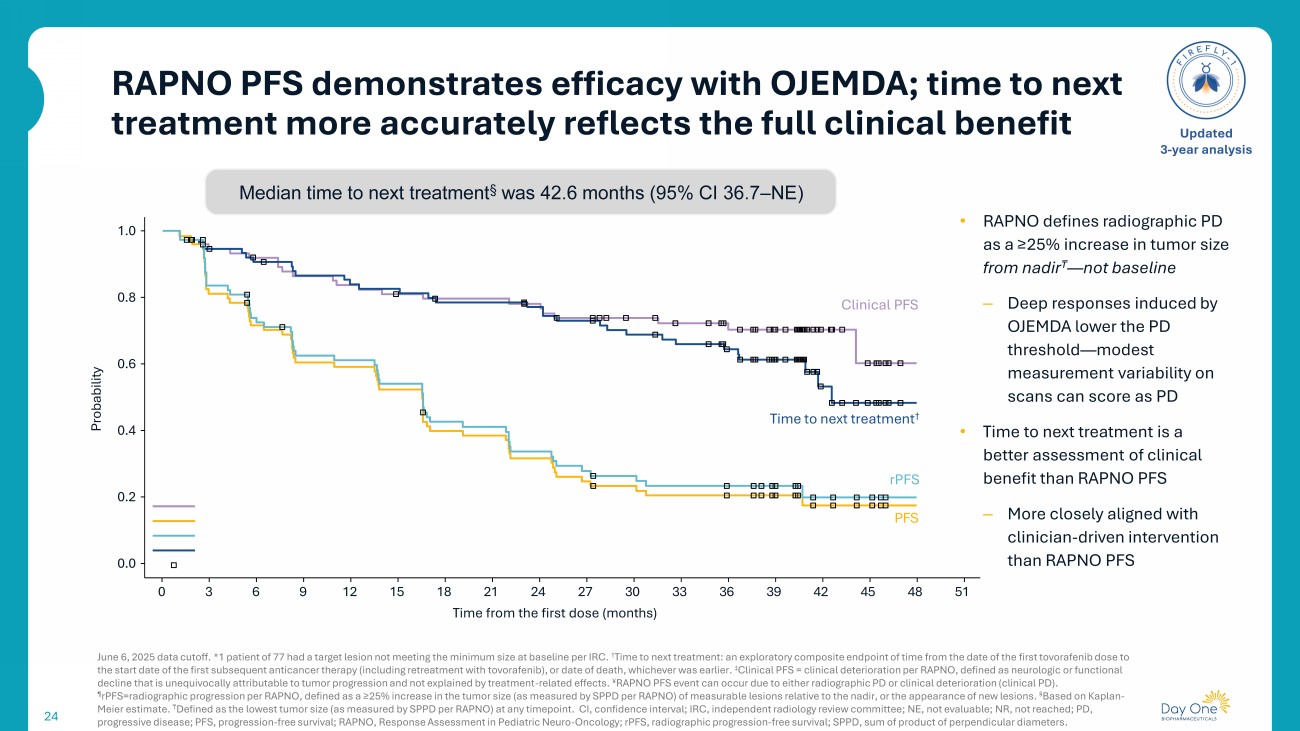
RAPNO PFS demonstrates efficacy with OJEMDA; time to next treatment more accurately reflects the full clinical benefit 24 June 6, 2025 data cutoff. *1 patient of 77 had a target lesion not meeting the minimum size at baseline per IRC. † T ime to next treatment : an exploratory composite endpoint of time from the date of the first tovorafenib dose to the start date of the first subsequent anticancer therapy (including retreatment with tovorafenib ), or date of death, whichever was earlier . ‡ Clinical PFS = clinical deterioration per RAPNO, defined as neurologic or functional decline that is unequivocally attributable to tumor progression and not explained by treatment - related effects. ¥ RAPNO PFS event can occur due to either radiographic PD or clinical deterioration (clinical PD). ¶ rPFS =radiographic progression per RAPNO, defined as a ≥25% increase in the tumor size (as measured by SPPD per RAPNO) of measurab le lesions relative to the nadir, or the appearance of new lesions. § Based on Kaplan - Meier estimate. ₸ Defined as the lowest tumor size (as measured by SPPD per RAPNO) at any timepoint. CI, confidence interval; IRC, independent radiology review committee ; NE, not evaluable; NR, not reached; PD, progressive disease; PFS, progression - free survival; RAPNO, Response Assessment in Pediatric Neuro - Oncology ; rPFS , radiographic progression - free survival; SPPD, sum of product of perpendicular diameters . Updated 3 - year analysis 1.0 0.0 0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45 48 51 Time from the first dose (months) 0.8 0.6 0.4 0.2 Probability Clinical PFS rPFS PFS T ime to next treatment † Median time to next treatment § was 42.6 months (95% CI 36.7 – NE) • RAPNO defines radiographic PD as a ≥25% increase in tumor size from nadir ₸ — not baseline – Deep responses induced by OJEMDA lower the PD threshold — modest measurement variability on scans can score as PD • Time to next treatment is a better assessment of clinical benefit than RAPNO PFS – More closely aligned with clinician - driven intervention than RAPNO PFS
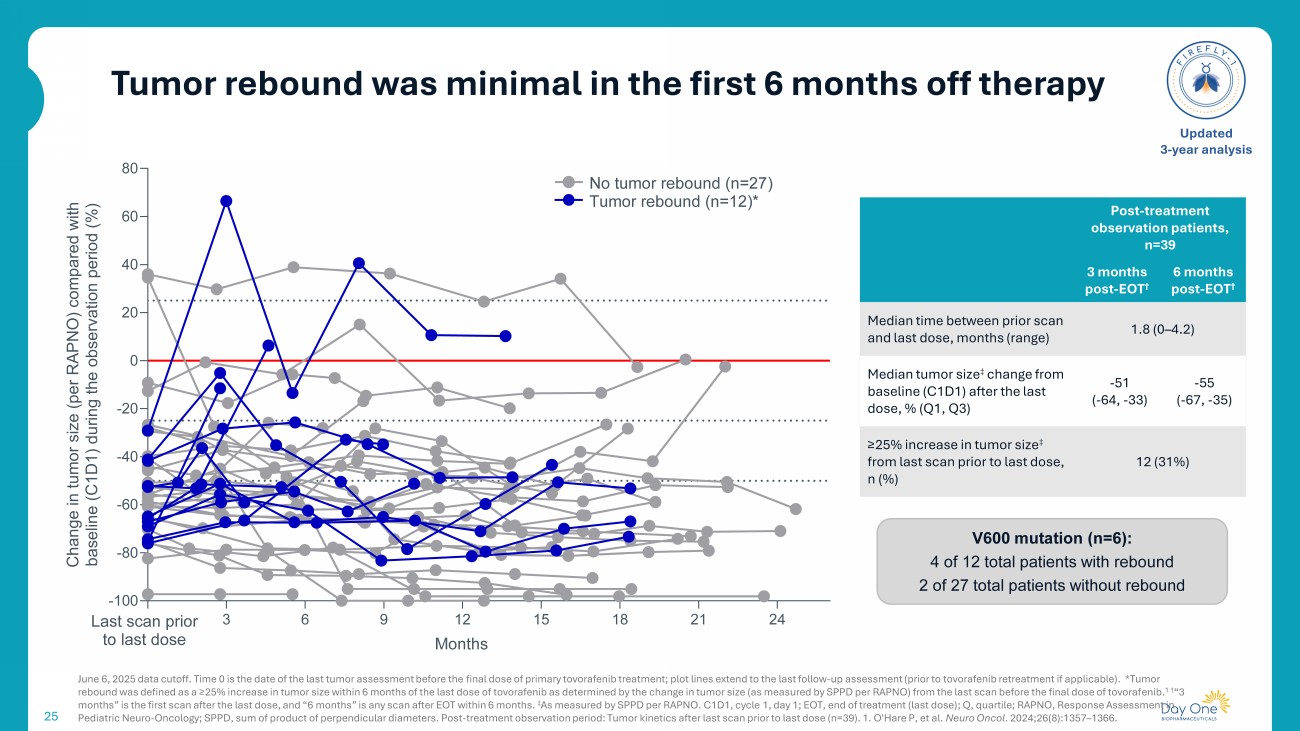
Tumor rebound was minimal in the first 6 months off therapy 25 June 6, 2025 data cutoff. Time 0 is the date of the last tumor assessment before the final dose of primary tovorafenib treatment; plot lines extend to the last follow - up assessment (prior to tovorafenib retreatment if applicable). *Tumor rebound was defined as a ≥25% increase in tumor size within 6 months of the last dose of tovorafenib as determined by the change in tumor size (as measured by SPPD per RAPNO) from the last scan before the final dose of tovoraf en ib. 1 † “3 months ” is the first scan after the last dose, and “6 months” is any scan after EOT within 6 months. ‡ As measured by SPPD per RAPNO. C1D1, cycle 1, day 1; EOT, end of treatment (last dose) ; Q, quartile; RAPNO, Response Assessment in Pediatric Neuro - Oncology; SPPD, sum of product of perpendicular diameters. Post - treatment observation period: Tumor kinetics aft er last scan prior to last dose (n=39). 1. O'Hare P, et al. Neuro Oncol . 2024;26(8):1357 – 1366. Updated 3 - year analysis 0 3 6 9 12 15 18 21 24 -100 -80 -60 -40 -20 0 20 40 60 80 C h a n g e i n t u m o r s i z e ( p e r R A P N O ) c o m p a r e d w i t h b a s e l i n e ( C 1 D 1 ) d u r i n g t h e o b s e r v a t i o n p e r i o d ( % ) No tumor rebound (n=27) Tumor rebound (n=12)* Last scan prior to last dose Months Post - treatment observation patients, n=39 6 months post - EOT † 3 months post - EOT † 1.8 (0 – 4.2) Median time between prior scan and last dose, months (range) - 55 ( - 67, - 35) - 51 ( - 64, - 33) Median tumor size ‡ change from baseline (C1D1) after the last dose, % (Q1, Q3) 12 (31%) ≥25% increase in tumor size ‡ from last scan prior to last dose, n (%) V600 mutation (n=6): 4 of 12 total patients with rebound 2 of 27 total patients without rebound
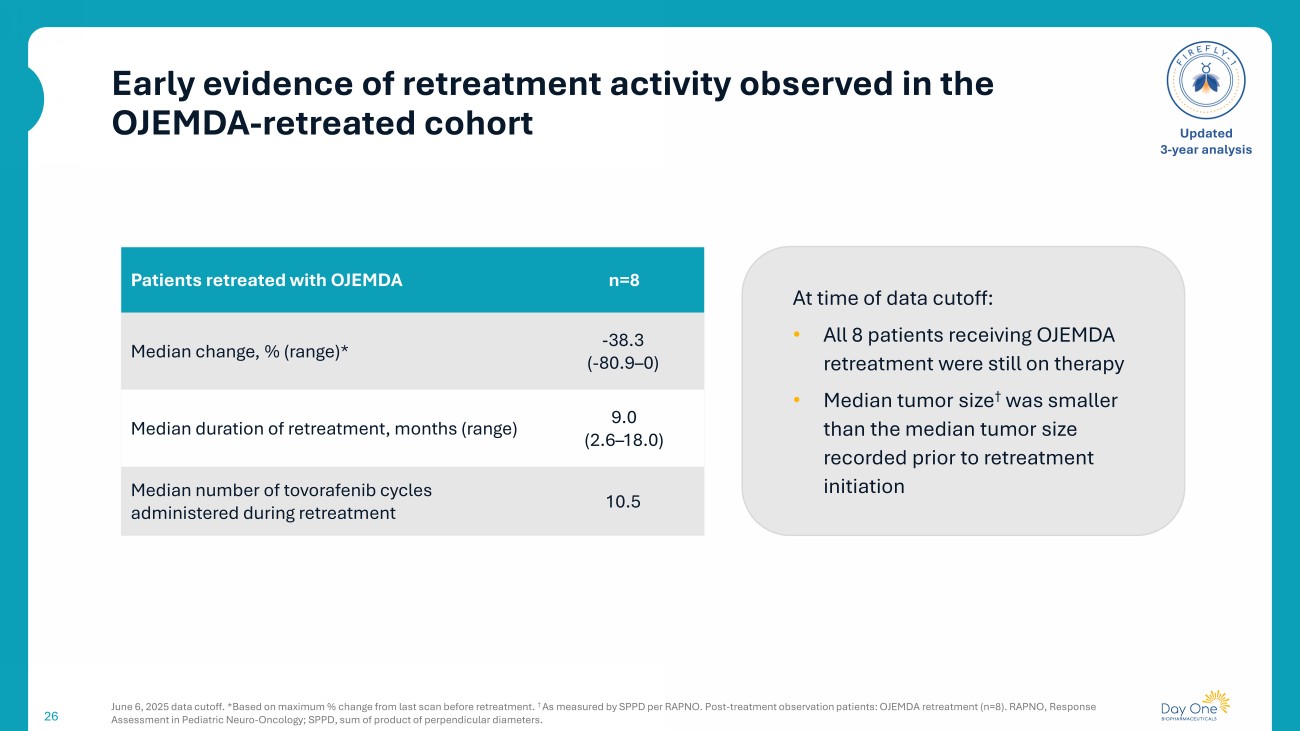
Early evidence of retreatment activity observed in the OJEMDA - retreated cohort 26 June 6, 2025 data cutoff. *Based on maximum % change from last scan before retreatment. † A s measured by SPPD per RAPNO. Post - treatment observation patients: OJEMDA retreatment (n=8). RAPNO, Response Assessment in Pediatric Neuro - Oncology; SPPD, sum of product of perpendicular diameters. Updated 3 - year analysis n=8 Patients retreated with OJEMDA - 38.3 ( - 80.9 – 0) Median change, % (range)* 9.0 (2.6 – 18.0) Median duration of retreatment, months (range) 10.5 Median number of tovorafenib cycles administered during retreatment At time of data cutoff: • All 8 patients receiving OJEMDA retreatment were still on therapy • Median tumor size † was smaller than the median tumor size recorded prior to retreatment initiation
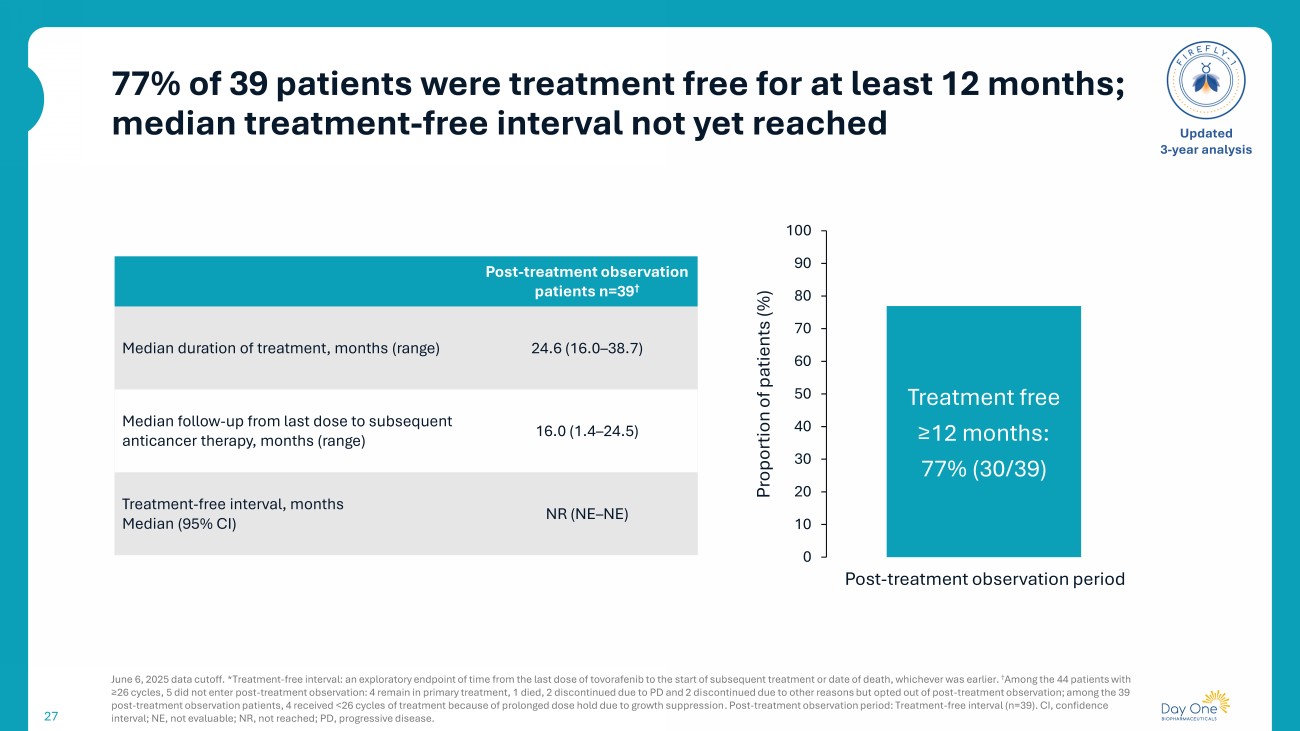
77% of 39 patients were treatment free for at least 12 months; median treatment - free interval not yet reached 27 June 6, 2025 data cutoff. * Treatment - free interval: an exploratory endpoint of time from the last dose of tovorafenib to the start of subsequent treatment or date of death, whichever was earlier. † Among the 44 patients with ≥26 cycles, 5 did not enter post - treatment observation: 4 remain in primary treatment, 1 died, 2 discontinued due to PD and 2 di scontinued due to other reasons but opted out of post - treatment observation; among the 39 post - treatment observation patients, 4 received <26 cycles of treatment because of prolonged dose hold due to growth suppression . Post - treatment observation period: Treatment - free interval (n=39). CI, confidence interval; NE, not evaluable; NR, not reached; PD, progressive disease. Updated 3 - year analysis Post - treatment observation patients n=39 † 24.6 (16.0 – 38.7) Median duration of treatment, months (range) 16.0 (1.4 – 24.5) Median follow - up from last dose to subsequent anticancer therapy, months (range) NR (NE – NE) Treatment - free interval, months Median (95% CI) Treatment free ≥12 months: 77 % (30/39) 0 10 20 30 40 50 60 70 80 90 100 Proportion of patients (%) Post - treatment observation period
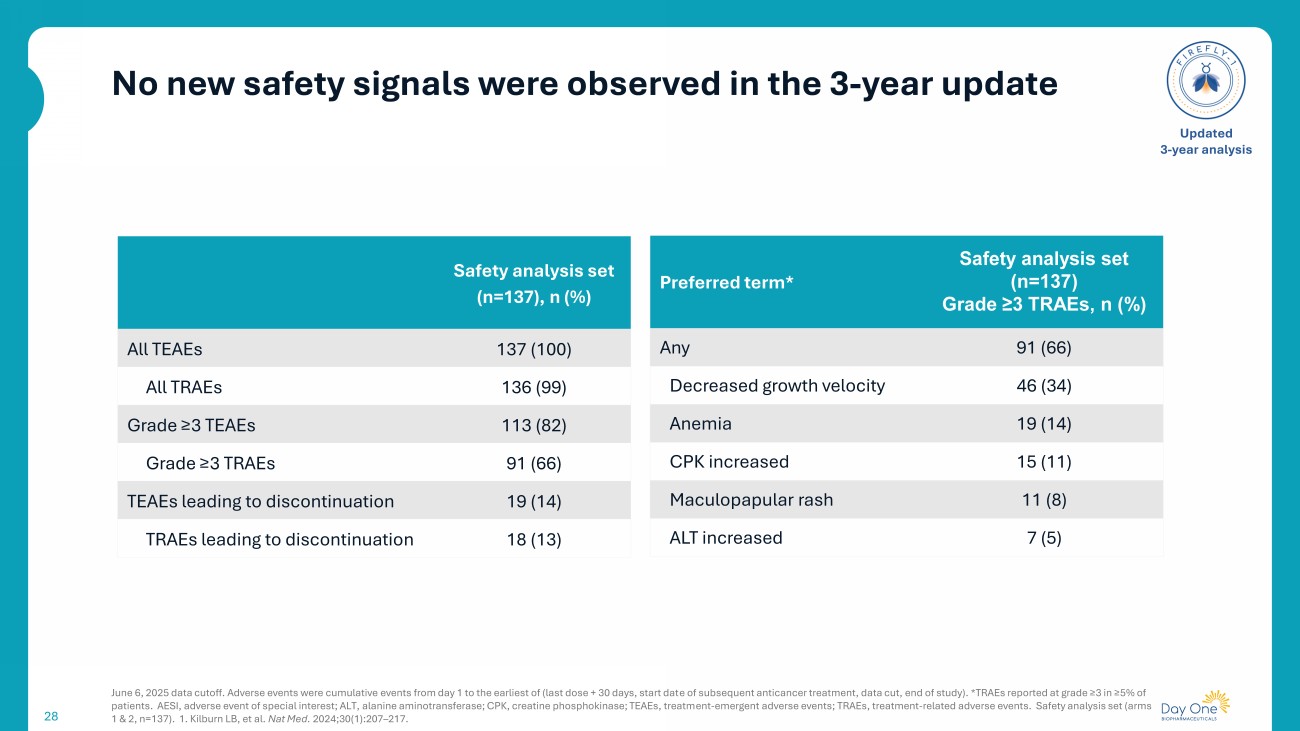
No new safety signals were observed in the 3 - year update 28 June 6, 2025 data cutoff. Adverse events were cumulative events from day 1 to the earliest of (last dose + 30 days, start dat e o f subsequent anticancer treatment , data cut, end of study ). *TRAEs reported at grade ≥3 in ≥5% of patients. AESI, adverse event of special interest; ALT, alanine aminotransferase; CPK, creatine phosphokinase; TEAEs, treatm ent - emergent adverse events; TRAEs, treatment - related adverse events. Safety analysis set (arms 1 & 2, n=137). 1. Kilburn LB, et al. Nat Med. 2024;30(1):207 – 217. Updated 3 - year analysis Safety analysis set (n=137), n (%) 137 (100) All TEAEs 136 (99) All TRAEs 113 (82) Grade ≥3 TEAEs 91 (66) Grade ≥3 TRAEs 19 (14) TEAEs leading to discontinuation 18 (13) TRAEs leading to discontinuation Safety analysis set (n=137) Grade ≥3 TRAEs, n (%) Preferred term* 91 (66) Any 46 (34) Decreased growth velocity 19 (14) Anemia 15 (11) CPK increased 11 (8) Maculopapular rash 7 (5) ALT increased

29 Pivotal Phase 3 trial of tovorafenib in front - line pLGG FIREFLY - 2 Bradon Living with pLGG since age 11
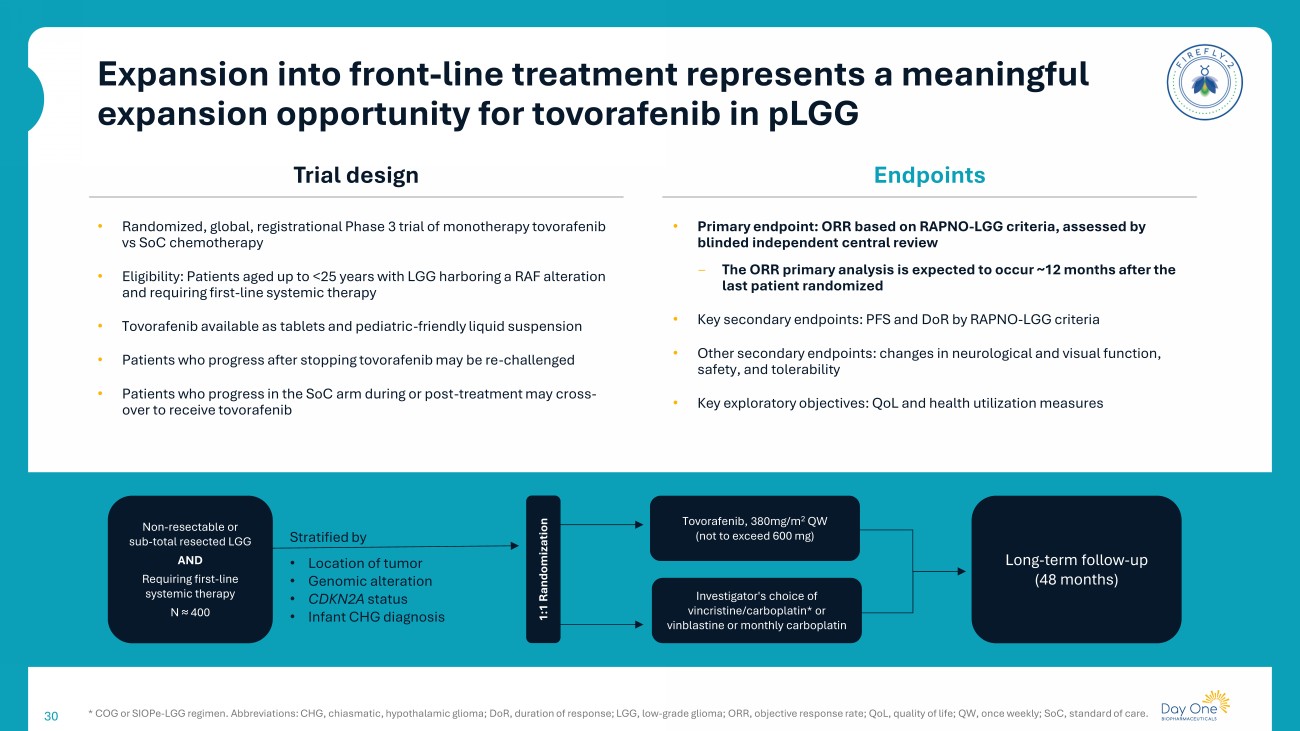
30 Trial design Endpoints • Randomized, global, registrational Phase 3 trial of monotherapy tovorafenib vs SoC chemotherapy • Eligibility: Patients aged up to <25 years with LGG harboring a RAF alteration and requiring first - line systemic therapy • Tovorafenib available as tablets and pediatric - friendly liquid suspension • Patients who progress after stopping tovorafenib may be re - challenged • Patients who progress in the SoC arm during or post - treatment may cross - over to receive tovorafenib • Primary endpoint: ORR based on RAPNO - LGG criteria, assessed by blinded independent central review ‒ The ORR primary analysis is expected to occur ~12 months after the last patient randomized • Key secondary endpoints: PFS and DoR by RAPNO - LGG criteria • Other secondary endpoints: changes in neurological and visual function, safety, and tolerability • Key exploratory objectives: QoL and health utilization measures Non - resectable or sub - total resected LGG AND Requiring first - line systemic therapy N ≈ 400 Stratified by • Location of tumor • Genomic alteration • CDKN2A status • Infant CHG diagnosis Tovorafenib , 380mg/m 2 QW (not to exceed 600 mg) Investigator's choice of vincristine/carboplatin* or vinblastine or monthly carboplatin Long - term follow - up (48 months) 1:1 Randomization * COG or SIOPe - LGG regimen. Abbreviations: CHG, chiasmatic, hypothalamic glioma; DoR , duration of response; LGG, low - grade glioma; ORR, objective response rate; QoL, quality of life; QW, once weekly; SoC, standar d of care. Expansion into front - line treatment represents a meaningful expansion opportunity for tovorafenib in pLGG

B7 - H4 - targeted antibody - drug conjugate (ADC) 31 Emi - Le

32 Emi - Le represents a transformational opportunity to address the unmet need for patients with adenoid cystic carcinoma (ACC) • ACC is a rare cancer, with an annual US incidence of ~1,300 patients 2 • Recurrent/metastatic ACC often presents with aggressive features; no approved therapeutic options exist 2 • B7 - H4 is highly and uniformly overexpressed in patients with recurrent/metastatic ACC 3 • B7 - H4 is also expressed in other adult and pediatric tumor types with high unmet need 4 • Emi - Le is an investigational B7 - H4 - directed ADC, utilizing a target - optimized molecular design and a proprietary linker - payload ( Dolasynthen ) 5 • Measurable anti - tumor activity observed in patients living with ACC - 1 and a well - defined safety profile support accelerated clinical development 1,6 Emi - Le ( Emiltatug Ledadotin ) 1 Potential First - in - Class B7 - H4 - targeted ADC opportunity in ACC 1 Hamilton EP et al. 2025 ASCO Annual Meeting; 2 Adenoid Cystic Carcinoma Research Foundation; 3 Mota Siquera J et al 2024 Modern Pathology; 4 Dawidowicz M et al 2024 Cancers; 5 Fessler et al 2023 Mol Cancer Ther ; 6 Hamilton E et al 2025 ESMO Breast Cancer.

PTK7 - targeted antibody - drug conjugate (ADC) 33 DAY301

Substantial development and commercial potential for DAY301 Novel ADC active in preclinical models, designed to maximize therapeutic window 34 PTK7: clinically - validated ADC target Anti - tumor activity of anti - PTK7 ADC demonstrated in Phase 1b trial of cofetuzumab pelidotin 1 DAY301: potential first - in - class asset High PTK7 expression in multiple adult and pediatric tumor indications First dose cohort cleared January 2025 1 Cho BC, et al. Ann Oncol. (34; Suppl 2): S460 - S461, 2023. DAY301: Next generation ADC targeting PTK7

35 Potential opportunity for a next - generation PTK7 ADC with improved therapeutic index • Clinical results for cofetuzumab pelidotin 1 demonstrated proof of concept for PTK7 - targeted ADCs • Cofetuzumab pelidotin activity seen in multiple tumor types: • Ovarian (Pt - resistant): ORR 27% (n=63) • TNBC: ORR 21% (n=29) • NSCLC: ORR 19% (n=31) • mDOR : 4.2 - 5.7m for Ovarian (Pt - resistant)/TNBC/NSCLC • mPFS : 1.5 - 2.9m for Ovarian (Pt - resistant)/TNBC/NSCLC • Aur0101 program limited by toxicity, resulting in reduced dose intensity and duration • A next generation product with optimized properties and a better therapeutic index may achieve greater clinical efficacy 1 Phase 1b study of PF - 06647020/ABBV - 647. PTK7: A clinically - validated ADC target

36 DAY301 has been designed to maximize therapeutic index and overcome limitations of prior programs • Tumor regression at tolerable doses seen in multiple preclinical models • Higher HNSTD in cyno toxicology studies; payload with known safety profile • High cell permeability / bystander effect; low efflux (not a P - gp substrate) • Novel, highly hydrophilic, cleavable linker • Moderate - to - high affinity antibody with favorable stability and developability profile • Drug - antibody - ratio (DAR) of 8, shown to be effective for other ADCs in solid tumors • IP: Composition of Matter patent term expected 2044, once issued 1) Damelin M, et al. A PTK7 - targeted antibody - drug conjugate reduces tumor - initiating cells and induces sustained tumor regressions. Sci Transl Med. 2017. HNSTD, Highest Non - Severely Toxic Dose; P - gp , P - glycoprotein. DAY301: Potential first - in - class asset
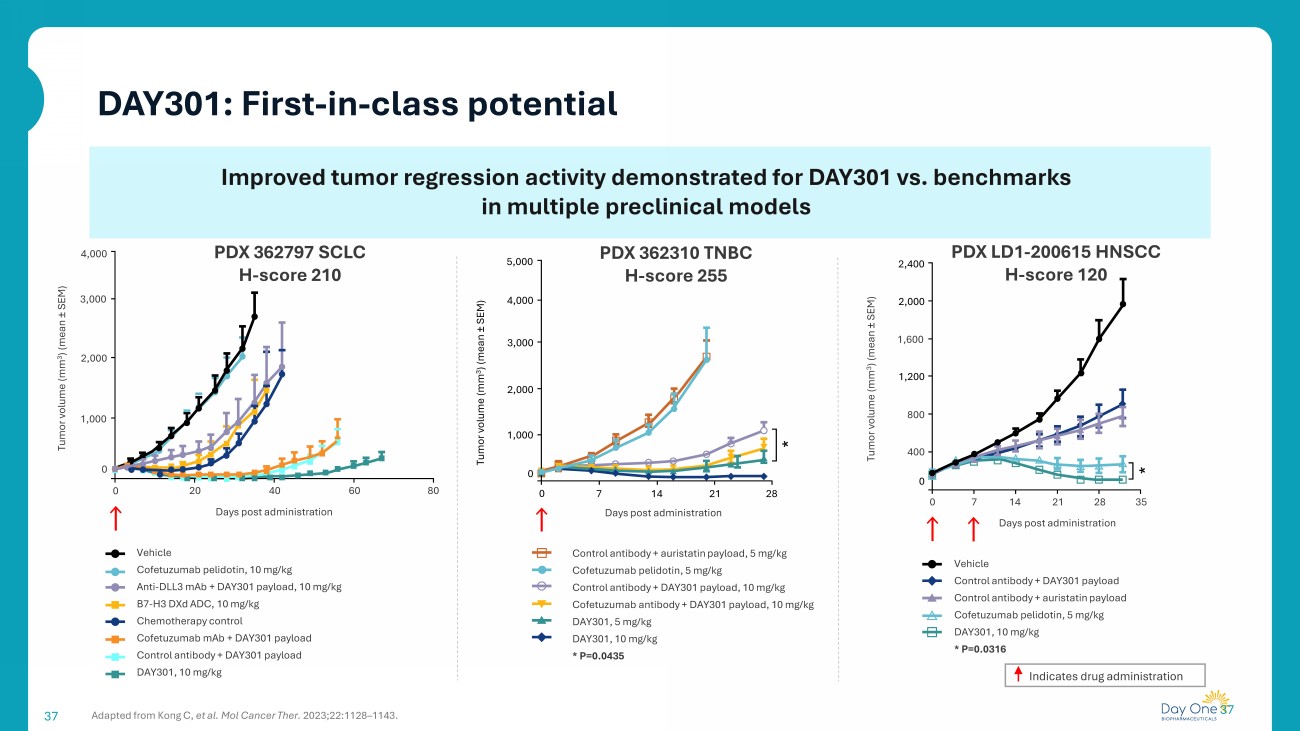
37 37 Improved tumor regression activity demonstrated for DAY301 vs. benchmarks in multiple preclinical models DAY301: First - in - class potential PDX LD1 - 200615 HNSCC H - score 120 Vehicle Control antibody + DAY301 payload Control antibody + auristatin payload Cofetuzumab pelidotin , 5 mg/kg DAY301, 10 mg/kg * P=0.0316 Days post administration * 0 7 14 21 28 35 Tumor volume (mm 3 ) (mean “ SEM) 2,400 0 2,000 1,600 1,200 800 400 Control antibody + auristatin payload, 5 mg/kg Cofetuzumab pelidotin , 5 mg/kg Control antibody + DAY301 payload, 10 mg/kg Cofetuzumab antibody + DAY301 payload, 10 mg/kg DAY301, 5 mg/kg DAY301, 10 mg/kg * P=0.0435 0 PDX 362310 TNBC H - score 255 4,000 3,000 2,000 1,000 0 7 14 21 28 * Days post administration Tumor volume (mm 3 ) (mean “ SEM) 0 Vehicle Cofetuzumab pelidotin , 10 mg/kg Anti - DLL3 mAb + DAY301 payload, 10 mg/kg B7 - H3 DXd ADC, 10 mg/kg Chemotherapy control Cofetuzumab mAb + DAY301 payload Control antibody + DAY301 payload DAY301, 10 mg/kg 20 40 60 80 0 3,000 2,000 1,000 Days post administration PDX 362797 SCLC H - score 210 Tumor volume (mm 3 ) (mean “ SEM) Adapted from Kong C, et al. Mol Cancer Ther. 2023;22:1128 – 1143. Indicates drug administration 4,000 5,000
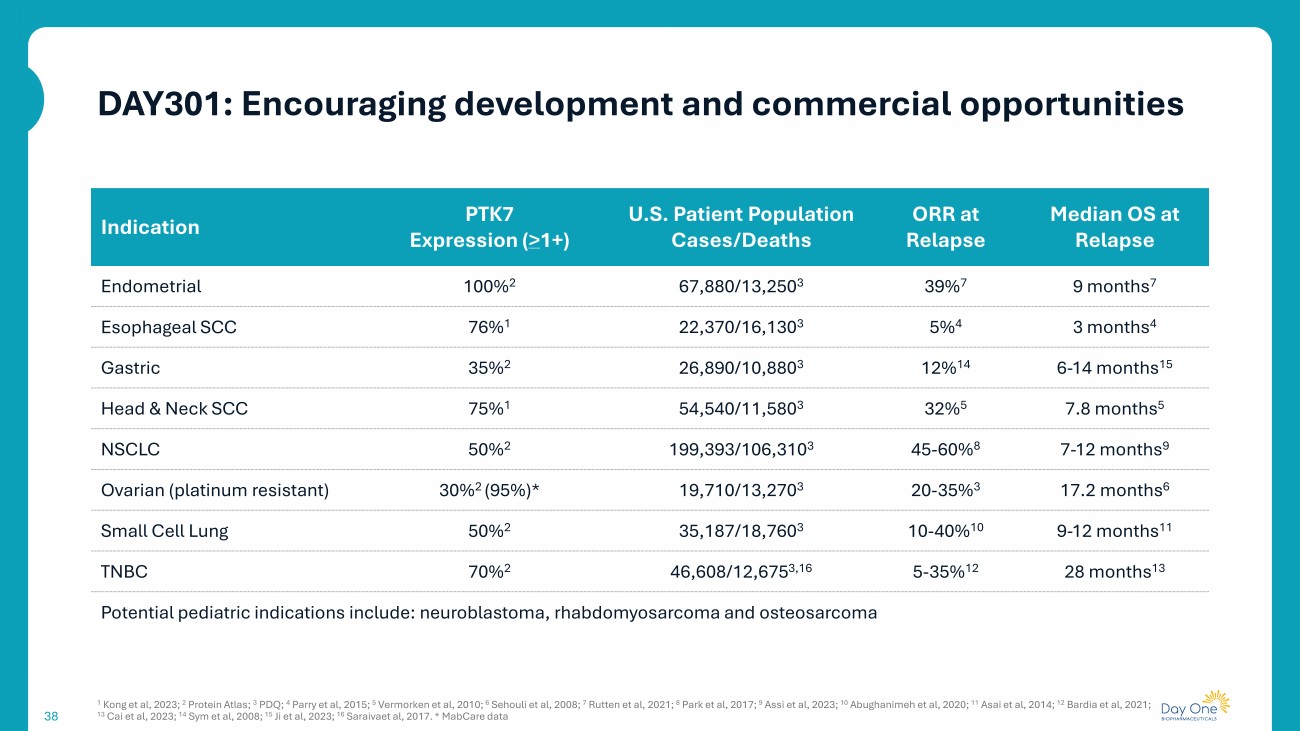
38 Median OS at Relapse ORR at Relapse U.S. Patient Population Cases/Deaths PTK7 Expression ( > 1+) Indication 9 months 7 39% 7 67,880/13,250 3 100% 2 Endometrial 3 months 4 5% 4 22,370/16,130 3 76% 1 Esophageal SCC 6 - 14 months 15 12% 14 26,890/10,880 3 35% 2 Gastric 7.8 months 5 32% 5 54,540/11,580 3 75% 1 Head & Neck SCC 7 - 12 months 9 45 - 60% 8 199,393/106,310 3 50% 2 NSCLC 17.2 months 6 20 - 35% 3 19,710/13,270 3 30% 2 (95%)* Ovarian (platinum resistant) 9 - 12 months 11 10 - 40% 10 35,187/18,760 3 50% 2 Small Cell Lung 28 months 13 5 - 35% 12 46,608/12,675 3,16 70% 2 TNBC Potential pediatric indications include: neuroblastoma, rhabdomyosarcoma and osteosarcoma 1 Kong et al, 2023; 2 Protein Atlas; 3 PDQ; 4 Parry et al, 2015; 5 Vermorken et al, 2010; 6 Sehouli et al, 2008; 7 Rutten et al, 2021; 8 Park et al, 2017; 9 Assi et al, 2023; 10 Abughanimeh et al, 2020; 11 Asai et al, 2014; 12 Bardia et al, 2021; 13 Cai et al, 2023; 14 Sym et al, 2008; 15 Ji et al, 2023; 16 Saraivaet al, 2017. * MabCare data DAY301: Encouraging development and commercial opportunities
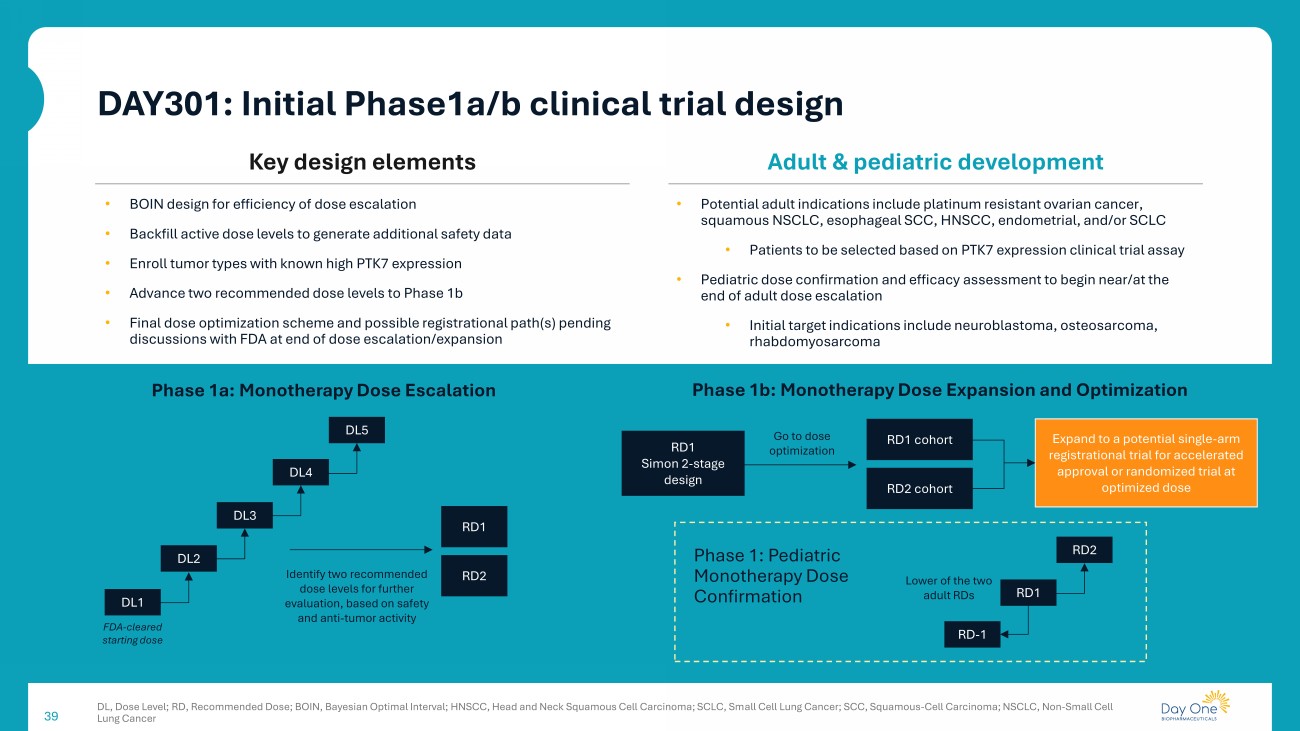
39 Phase 1a: Monotherapy Dose Escalation FDA - cleared starting dose DL5 RD1 RD2 Identify two recommended dose levels for further evaluation, based on safety and anti - tumor activity • BOIN design for efficiency of dose escalation • Backfill active dose levels to generate additional safety data • Enroll tumor types with known high PTK7 expression • Advance two recommended dose levels to Phase 1b • Final dose optimization scheme and possible registrational path(s) pending discussions with FDA at end of dose escalation/expansion RD1 Simon 2 - stage design Expand to a potential single - arm registrational trial for accelerated approval or randomized trial at optimized dose RD1 cohort RD2 cohort Go to dose optimization Phase 1b: Monotherapy Dose Expansion and Optimization Phase 1: Pediatric Monotherapy Dose Confirmation RD - 1 RD2 Lower of the two adult RDs • Potential adult indications include platinum resistant ovarian cancer, squamous NSCLC, esophageal SCC, HNSCC, endometrial, and/or SCLC • Patients to be selected based on PTK7 expression clinical trial assay • Pediatric dose confirmation and efficacy assessment to begin near/at the end of adult dose escalation • Initial target indications include neuroblastoma, osteosarcoma, rhabdomyosarcoma Key design elements Adult & pediatric development DL4 DL3 DL2 DL1 RD1 DL, Dose Level; RD, Recommended Dose; BOIN, Bayesian Optimal Interval; HNSCC, Head and Neck Squamous Cell Carcinoma; SCLC, Sm all Cell Lung Cancer; SCC, Squamous - Cell Carcinoma; NSCLC, Non - Small Cell Lung Cancer DAY301: Initial Phase1a/b clinical trial design

40 Summary
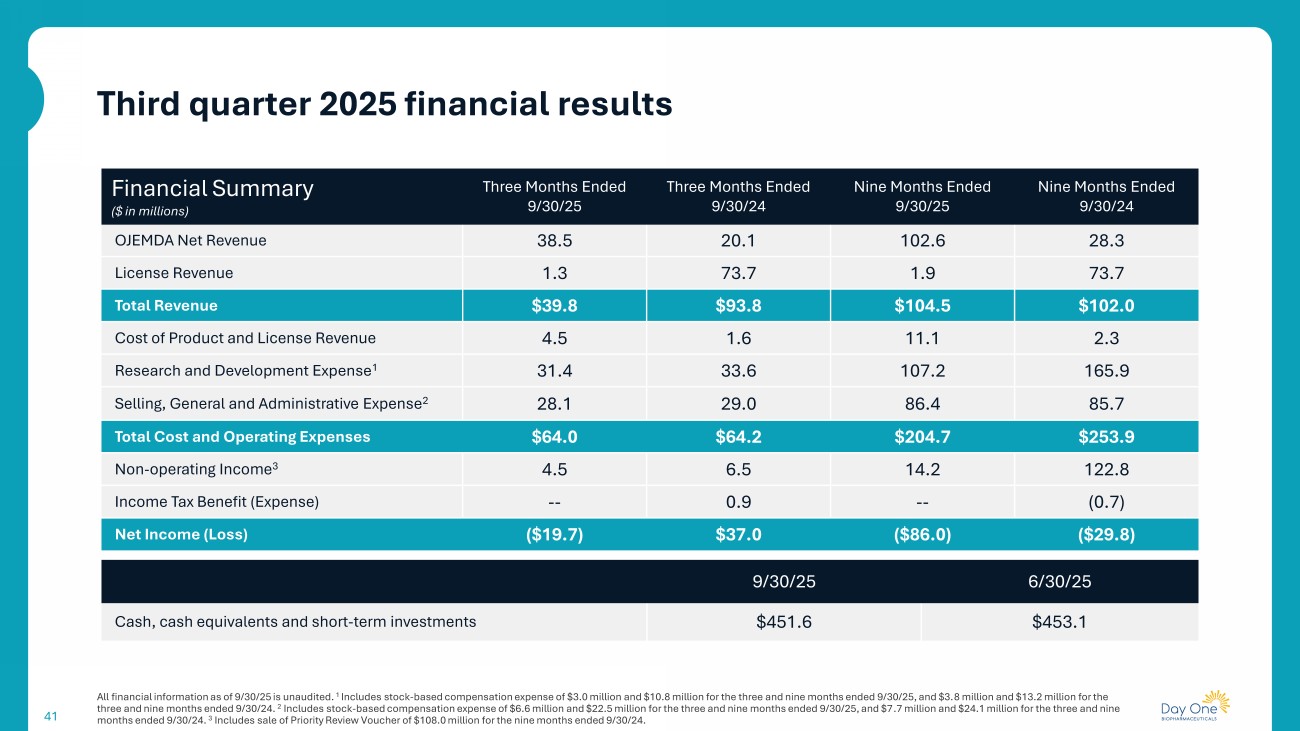
41 Third quarter 2025 financial results All financial information as of 9/30/25 is unaudited. 1 Includes stock - based compensation expense of $3.0 million and $10.8 million for the three and nine months ended 9/30/25, and $3. 8 million and $13.2 million for the three and nine months ended 9/30/24. 2 Includes stock - based compensation expense of $6.6 million and $22.5 million for the three and nine months ended 9/30/25, and $7 .7 million and $24.1 million for the three and nine months ended 9/30/24. 3 Includes sale of Priority Review Voucher of $108.0 million for the nine months ended 9/30/24. Nine Months Ended 9/30/24 Nine Months Ended 9/30/25 Three Months Ended 9/30/24 Three Months Ended 9/30/25 Financial Summary ($ in millions) 28.3 102.6 20.1 38.5 OJEMDA Net Revenue 73.7 1.9 73.7 1.3 License Revenue $102.0 $104.5 $93.8 $39.8 Total Revenue 2.3 11.1 1.6 4.5 Cost of Product and License Revenue 165.9 107.2 33.6 31.4 Research and Development Expense 1 85.7 86.4 29.0 28.1 Selling, General and Administrative Expense 2 $253.9 $204.7 $64.2 $64.0 Total Cost and Operating Expenses 122.8 14.2 6.5 4.5 Non - operating Income 3 (0.7) -- 0.9 -- Income Tax Benefit (Expense) ($29.8) ($86.0) $37.0 ($19.7) Net Income (Loss) 6/30/25 9/30/25 $453.1 $451.6 Cash, cash equivalents and short - term investments

42 Day One is well positioned for sustainable growth and long - term success 42 Drive OJEMDA revenue growth Execute on clinical development pipeline for FIREFLY - 2 and DAY301 Leverage our development and commercialization expertise to further expand our multiple asset portfolio Maintain strong capital position while investing in our pipeline

Appendix 43
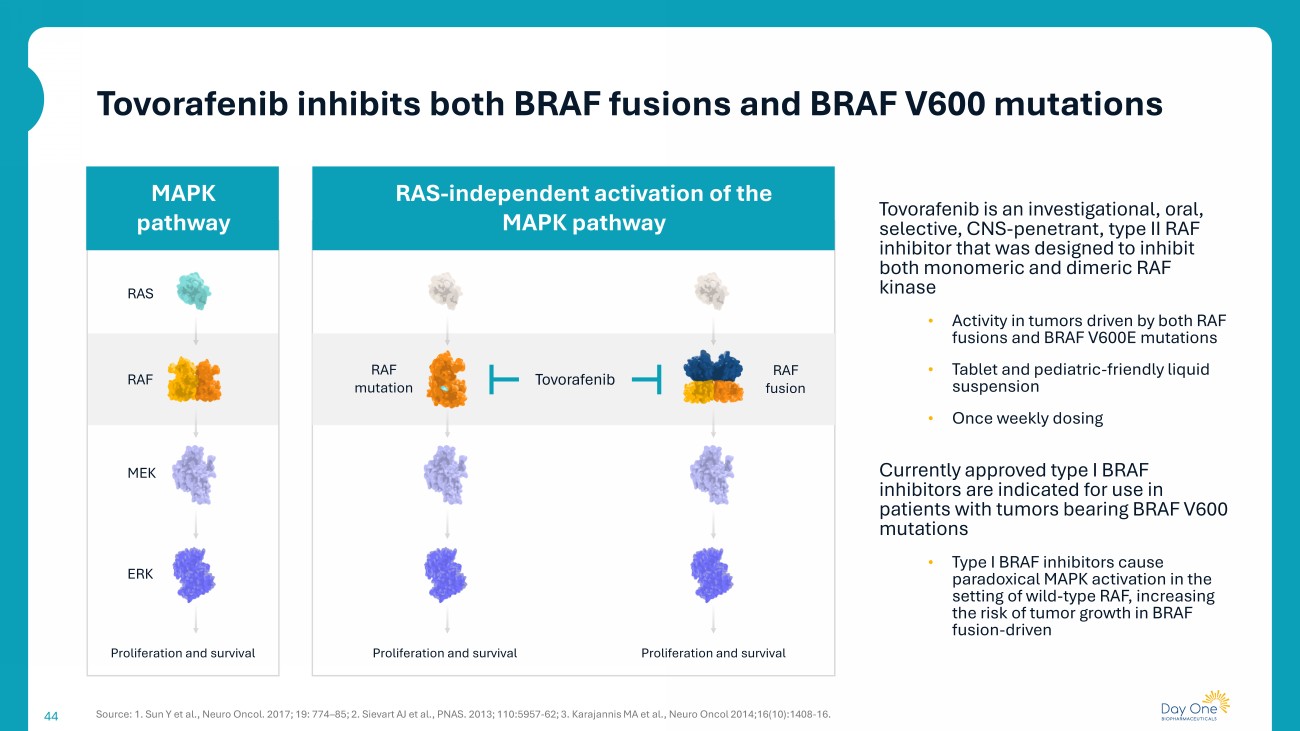
44 Tovorafenib is an investigational, oral, selective, CNS - penetrant, type II RAF inhibitor that was designed to inhibit both monomeric and dimeric RAF kinase • Activity in tumors driven by both RAF fusions and BRAF V600E mutations • Tablet and pediatric - friendly liquid suspension • Once weekly dosing Currently approved type I BRAF inhibitors are indicated for use in patients with tumors bearing BRAF V600 mutations • Type I BRAF inhibitors cause paradoxical MAPK activation in the setting of wild - type RAF, increasing the risk of tumor growth in BRAF fusion - driven RAS RAF MEK ERK Proliferation and survival RAF mutation RAF fusion Proliferation and survival Proliferation and survival Tovorafenib RAS - independent activation of the MAPK pathway MAPK pathway Source: 1. Sun Y et al., Neuro Oncol. 2017; 19: 774 – 85; 2. Sievart AJ et al., PNAS. 2013; 110:5957 - 62; 3. Karajannis MA et al., Neuro Oncol 2014;16(10):1408 - 16. Tovorafenib inhibits both BRAF fusions and BRAF V600 mutations