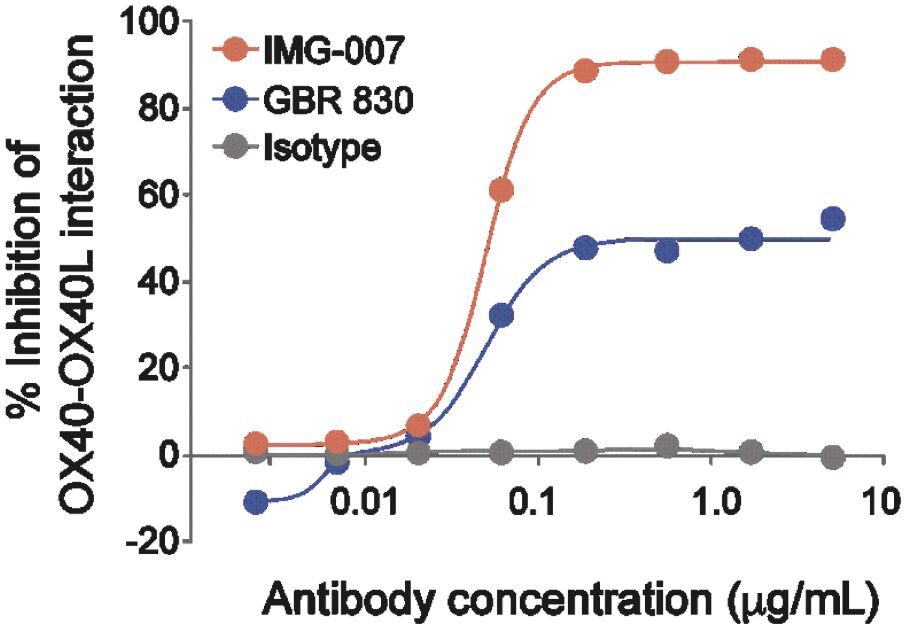
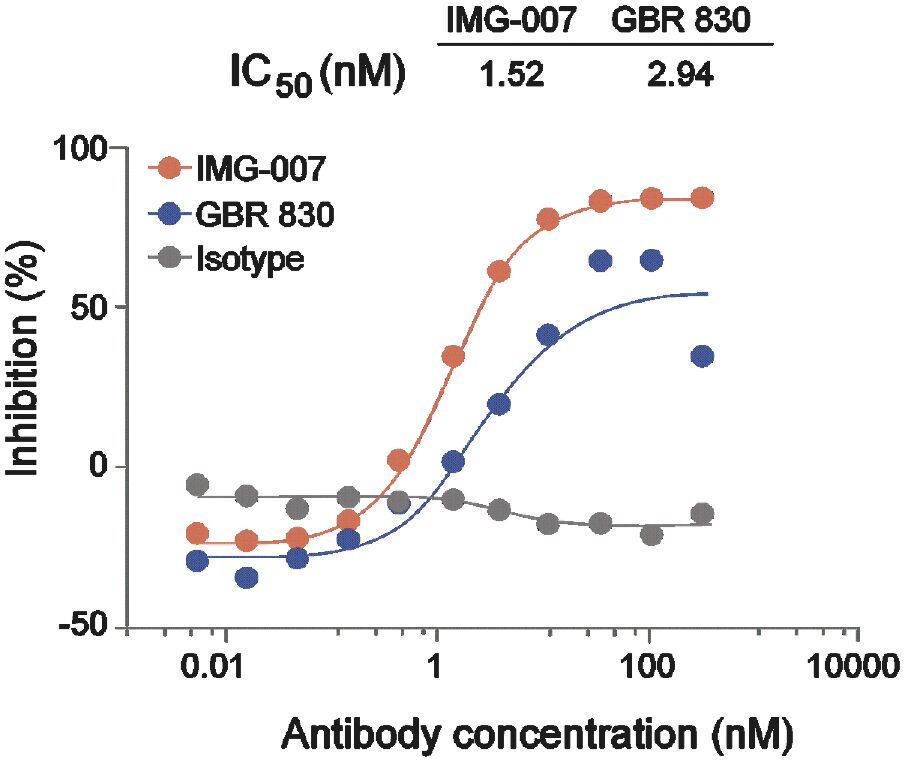
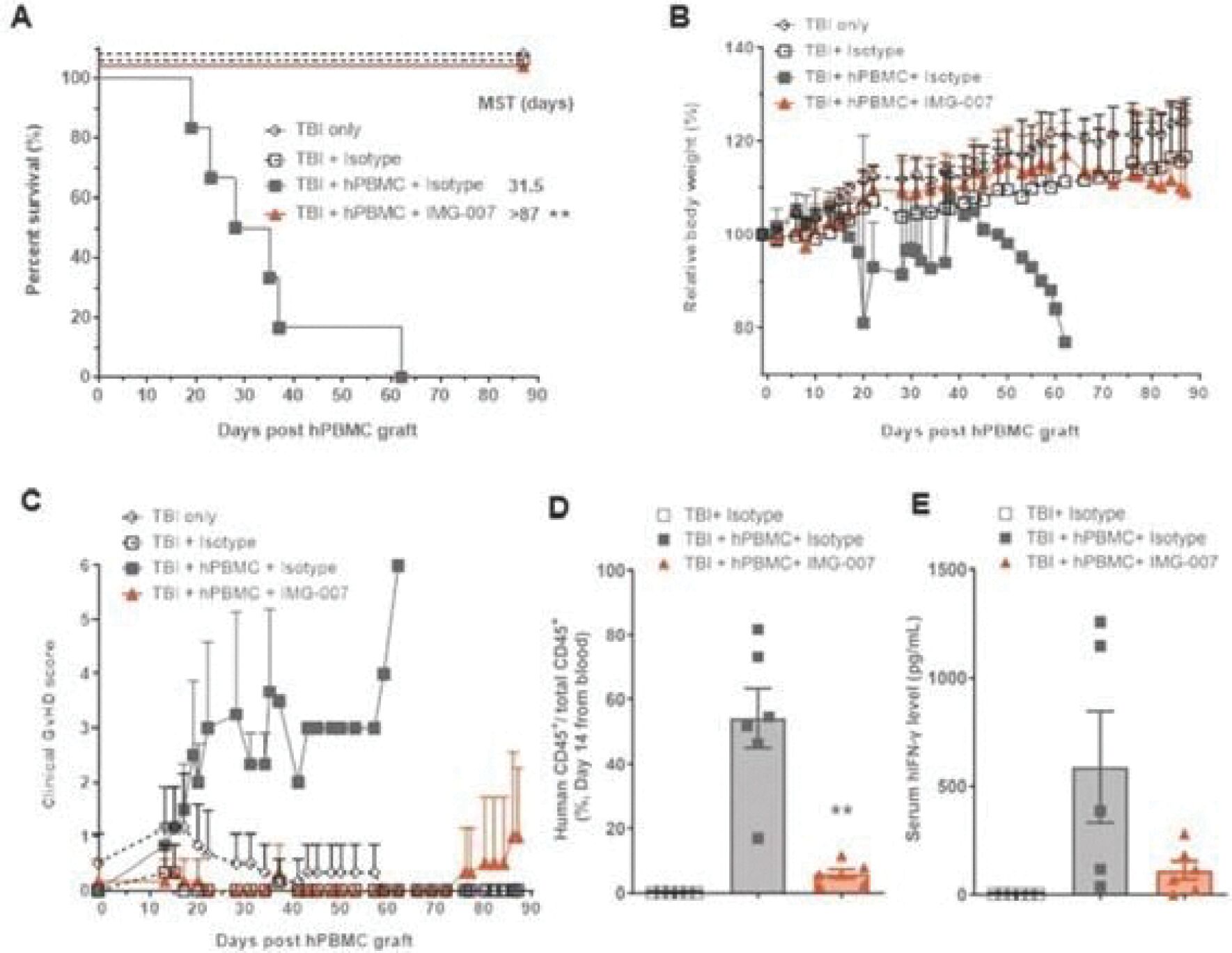
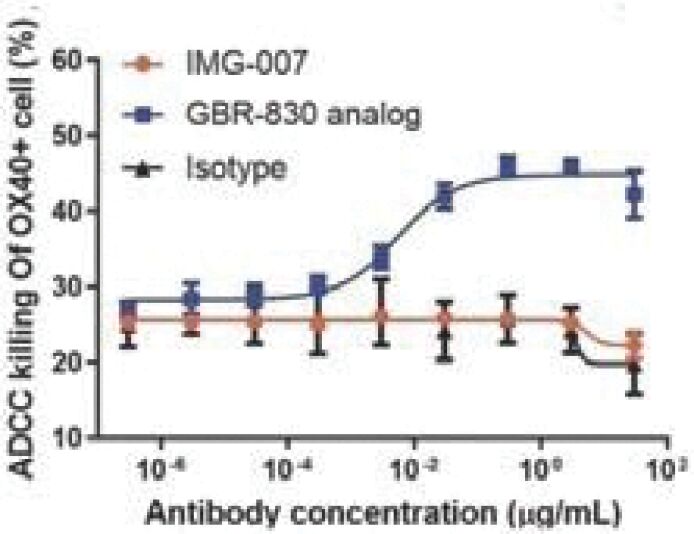
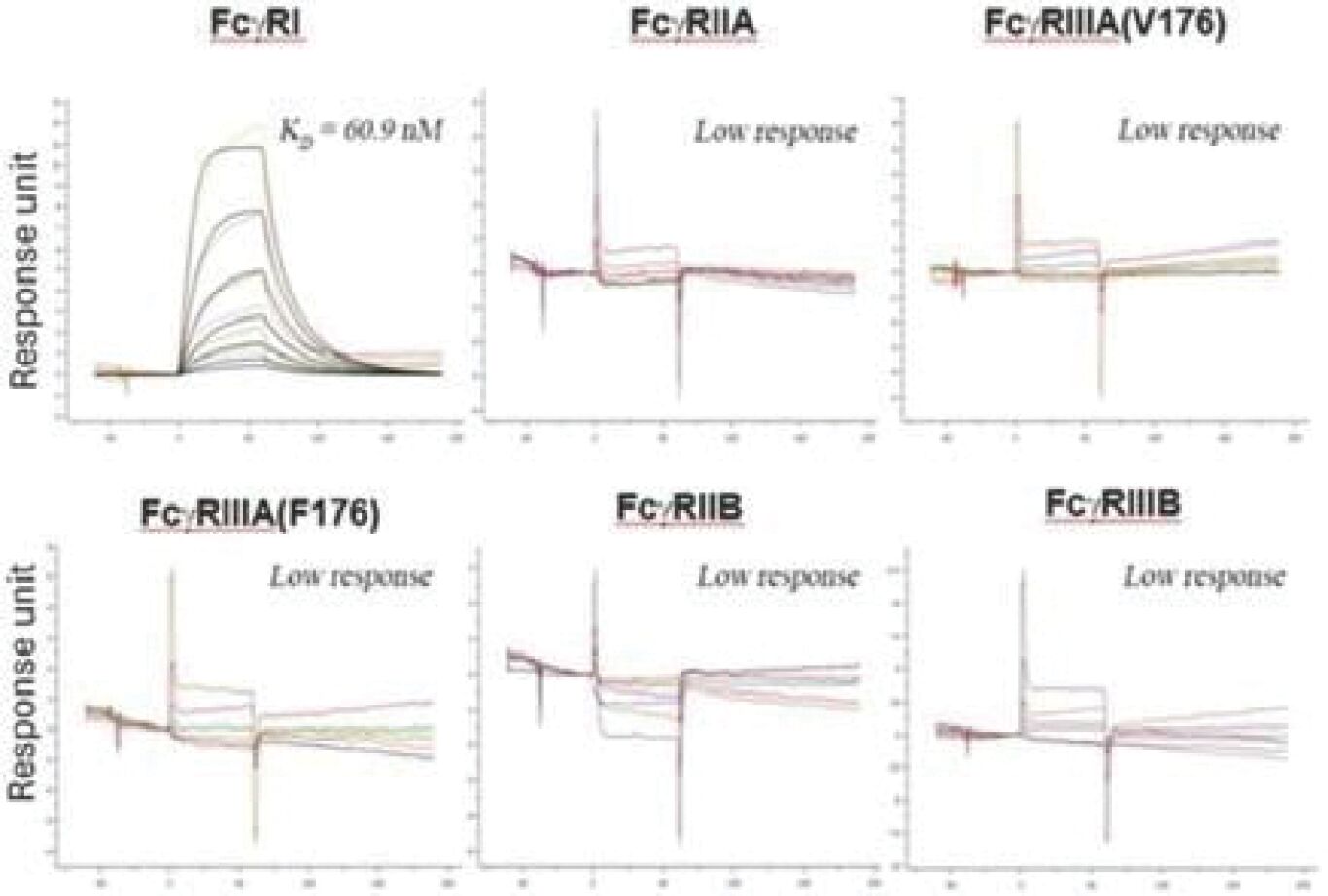
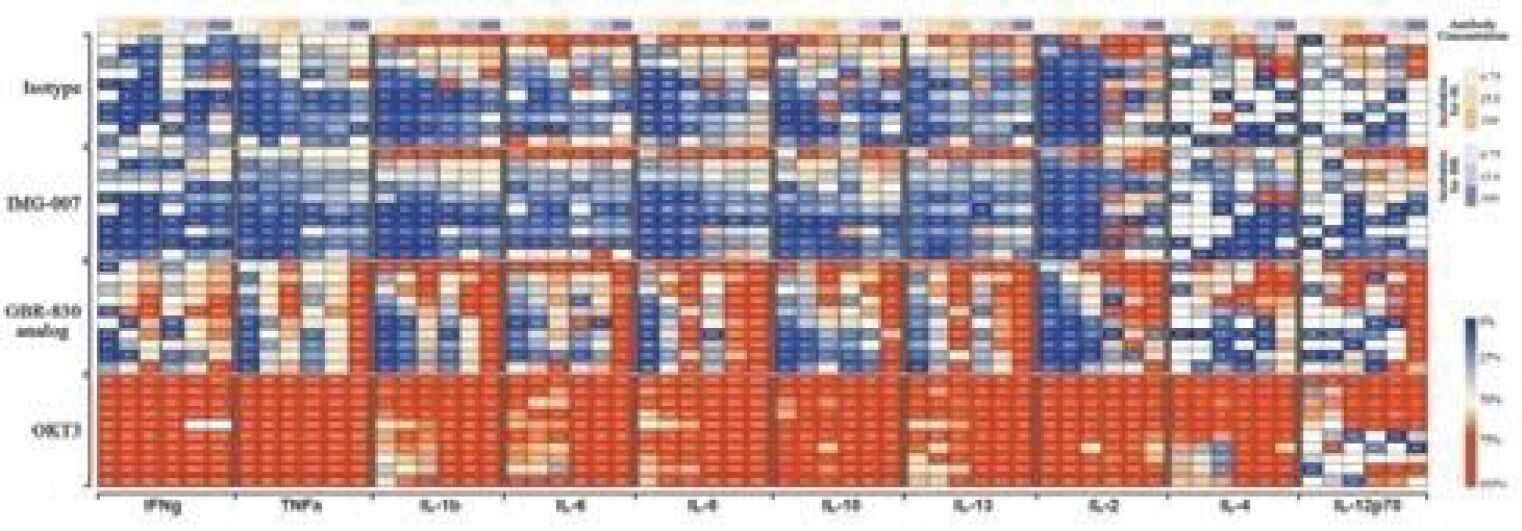
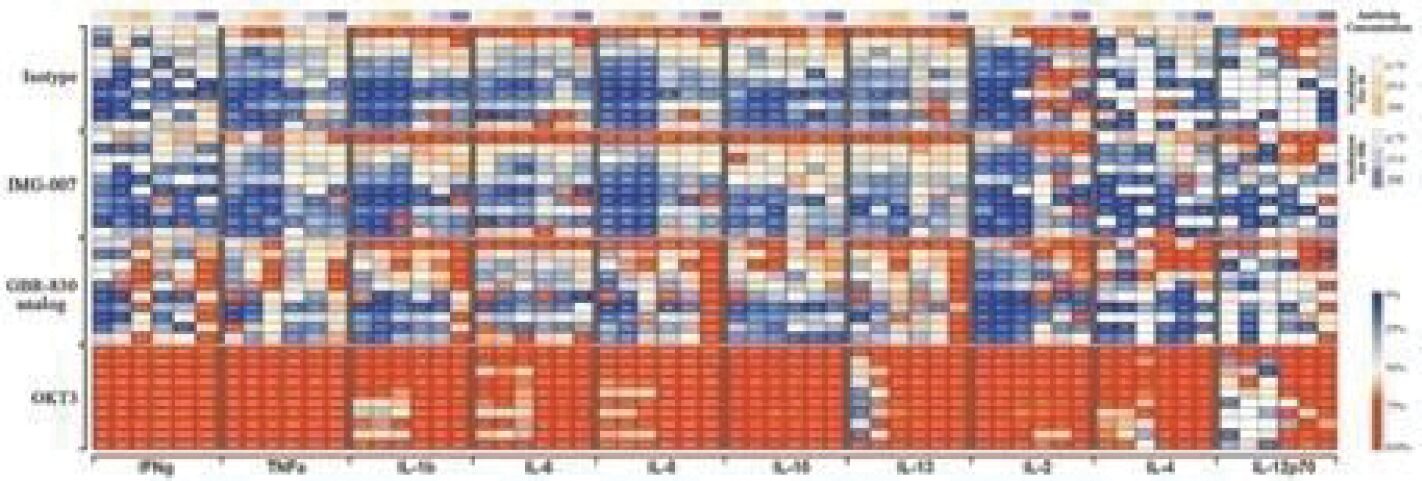
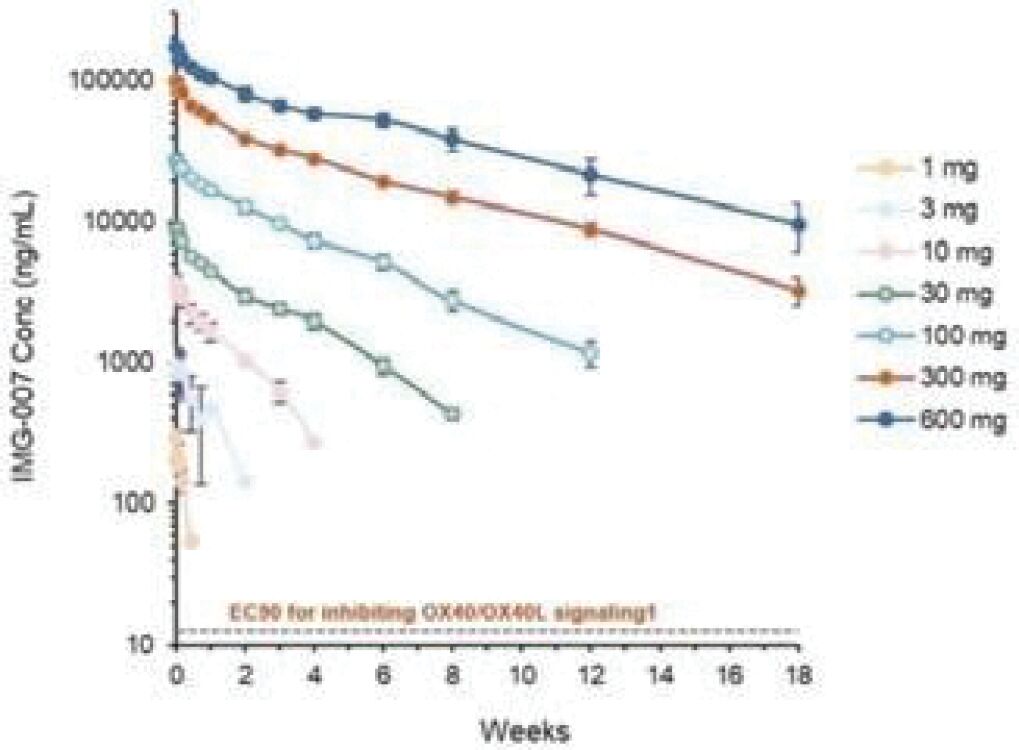
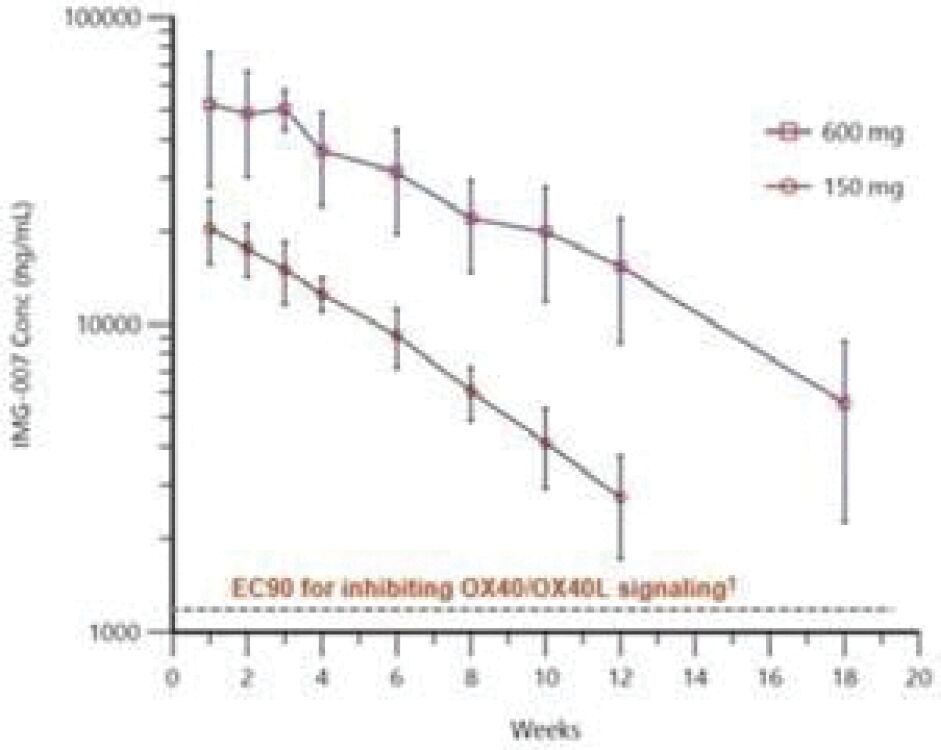
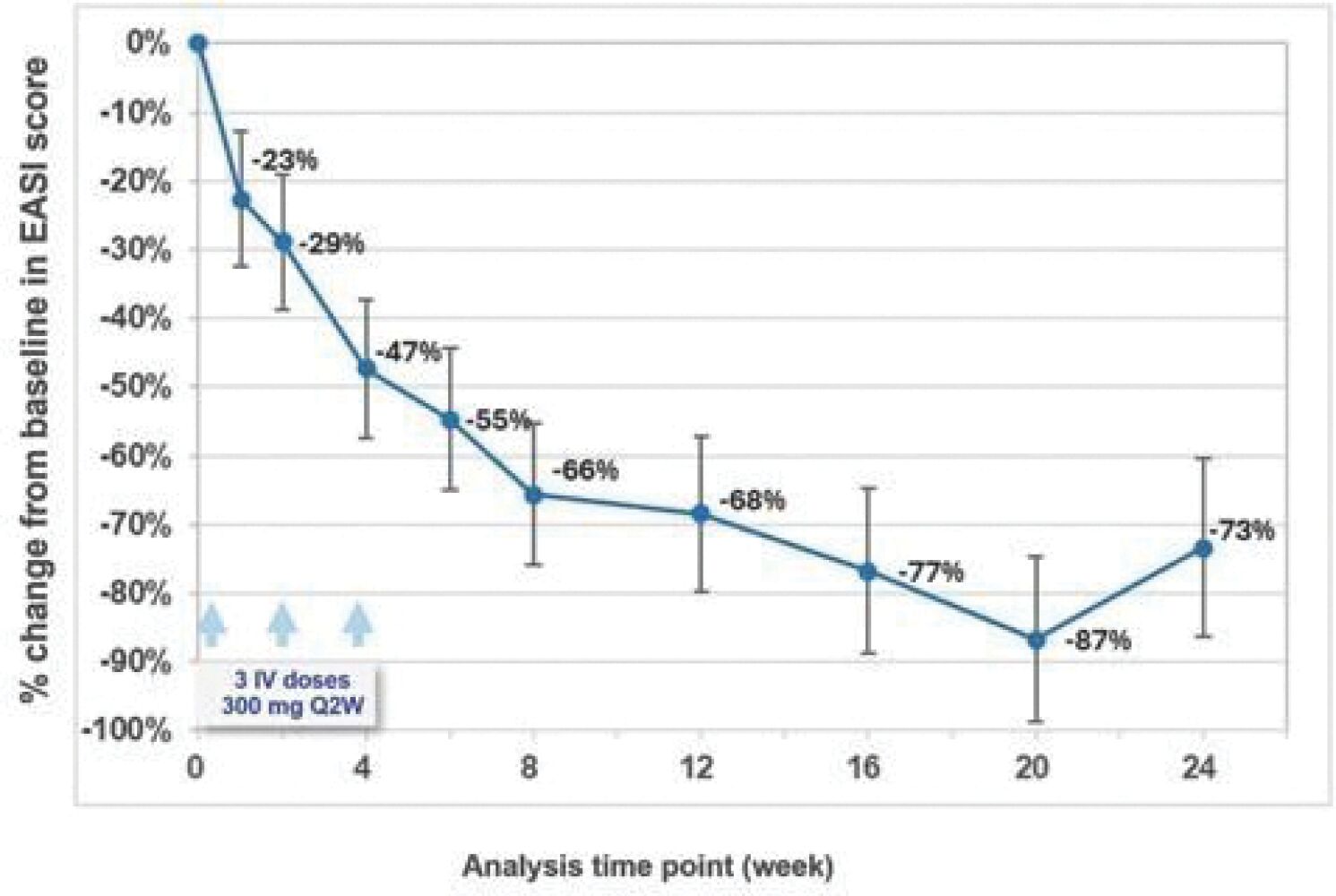
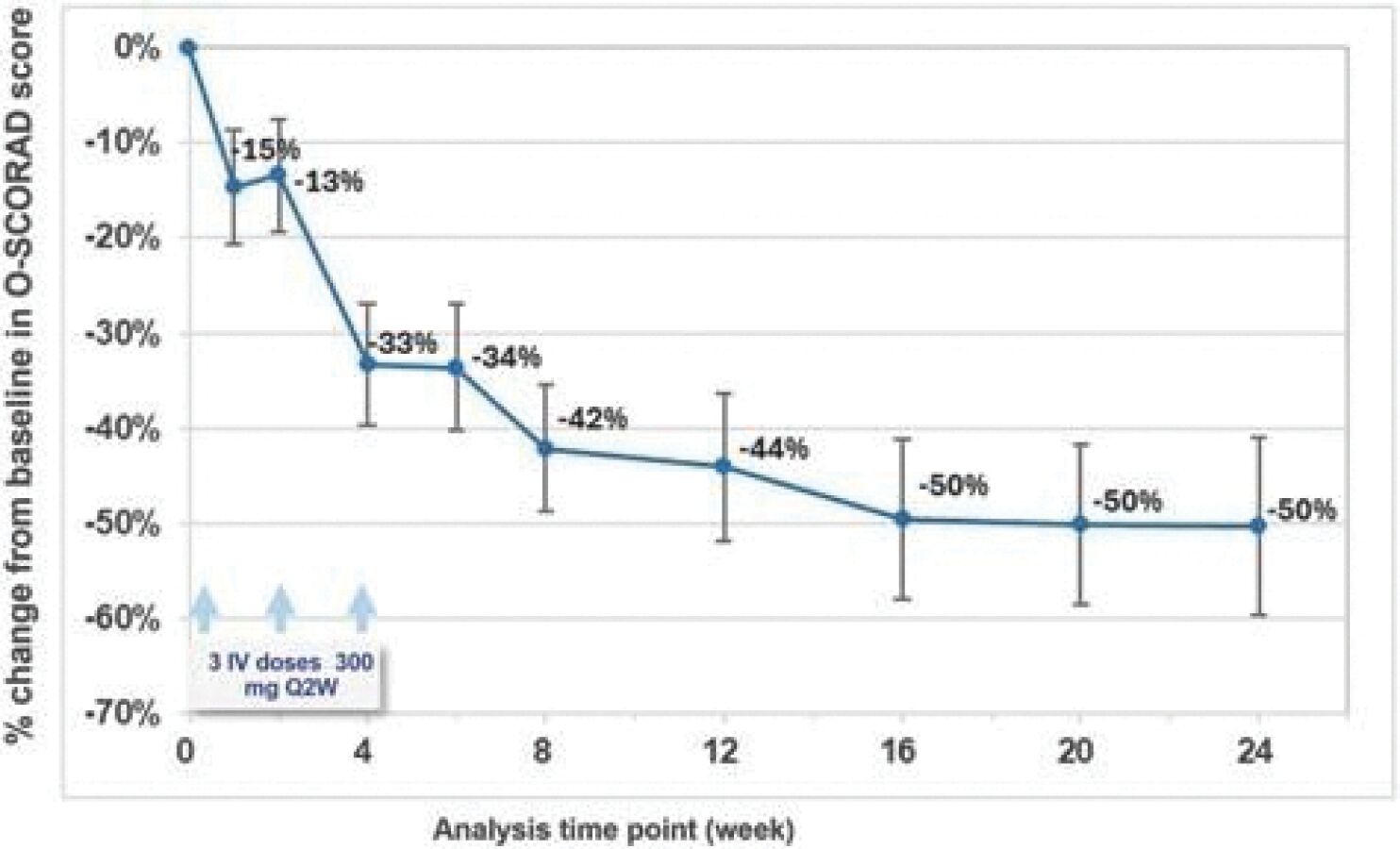
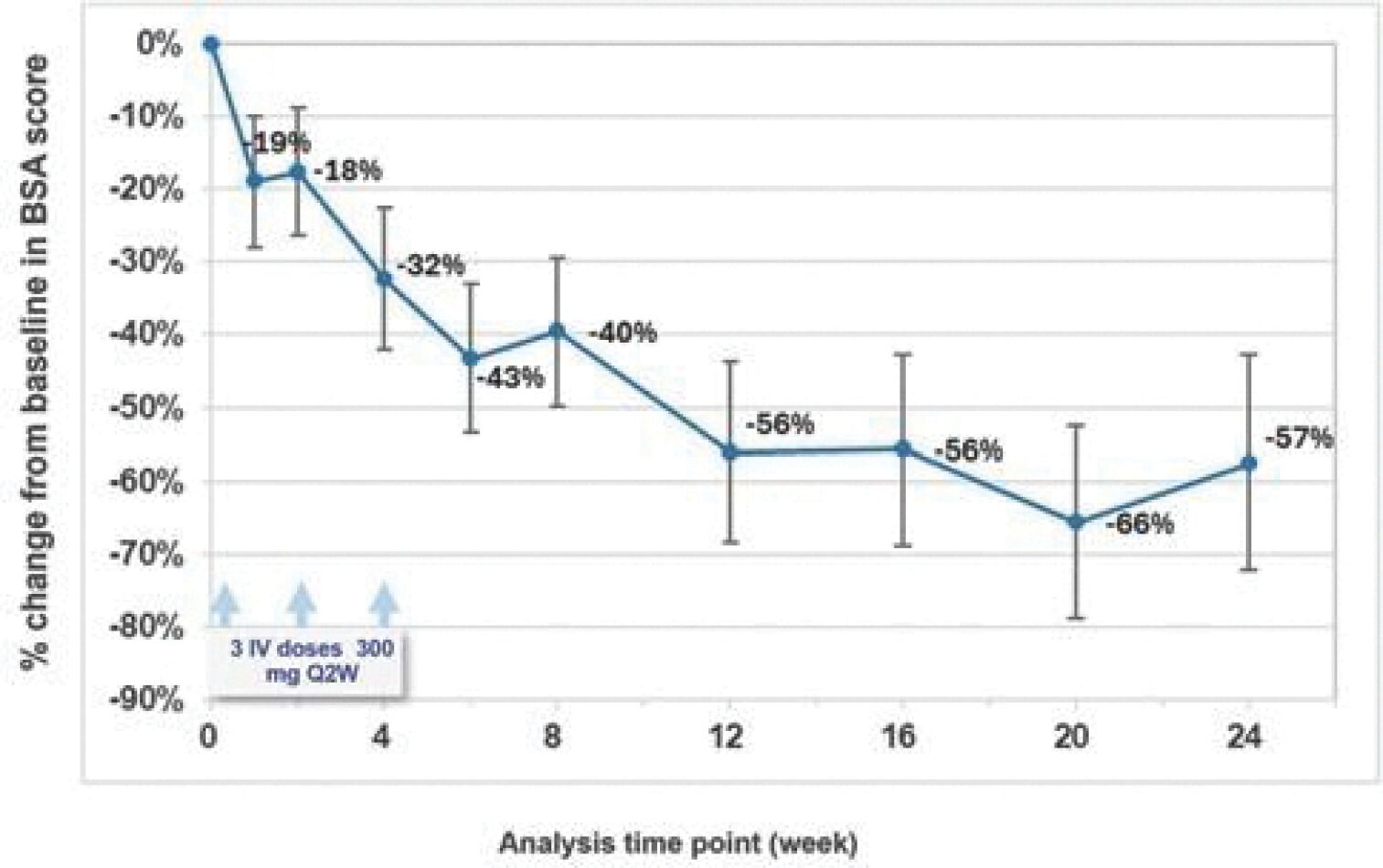
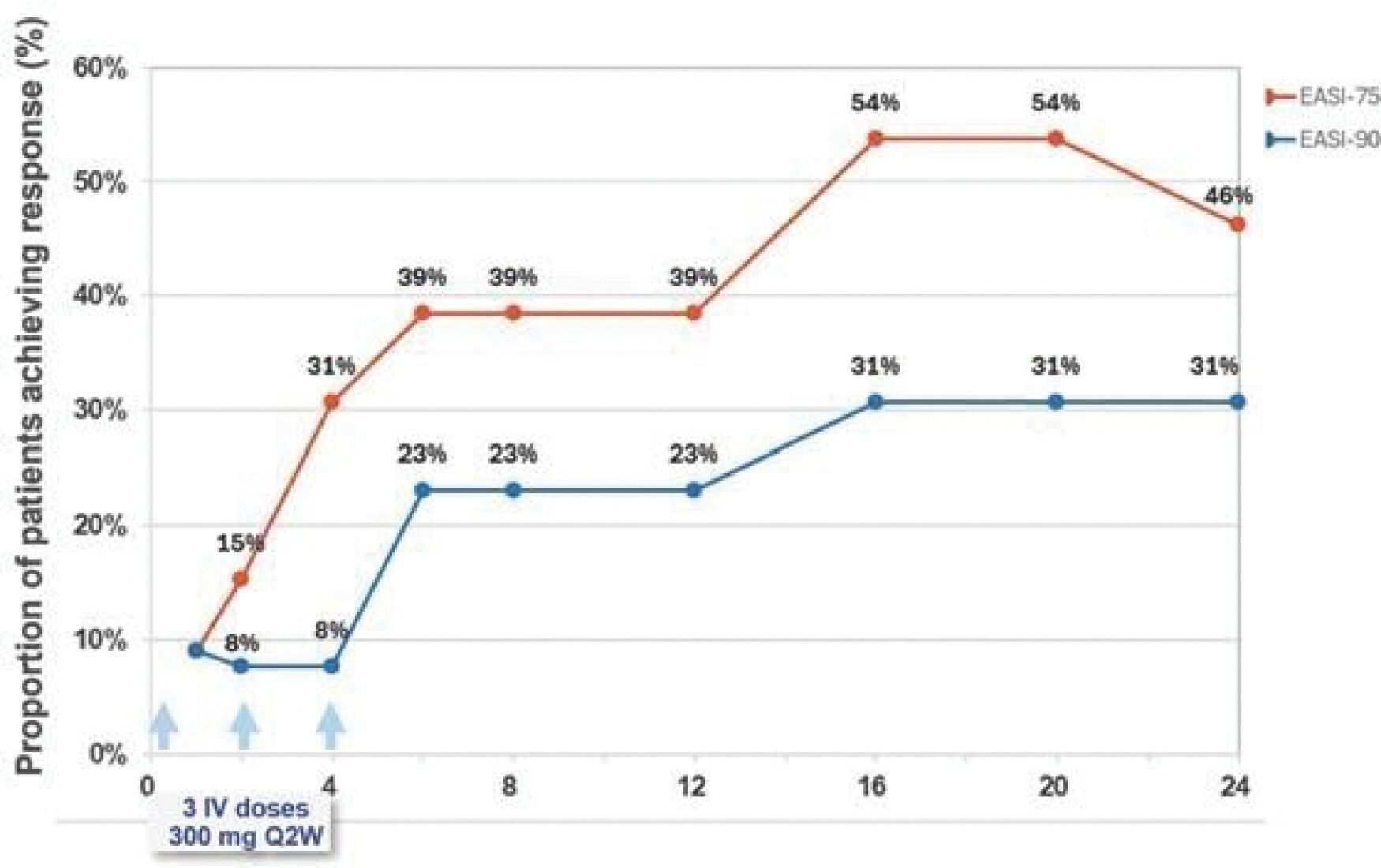
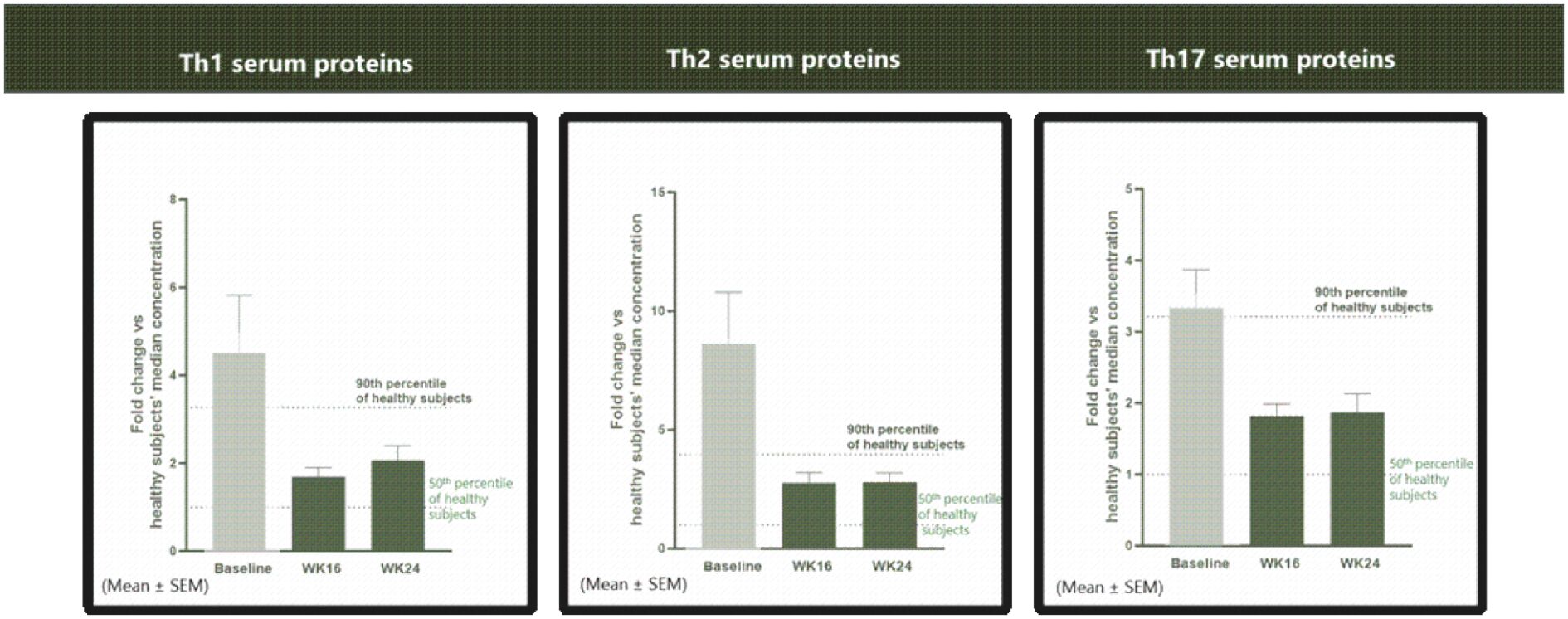
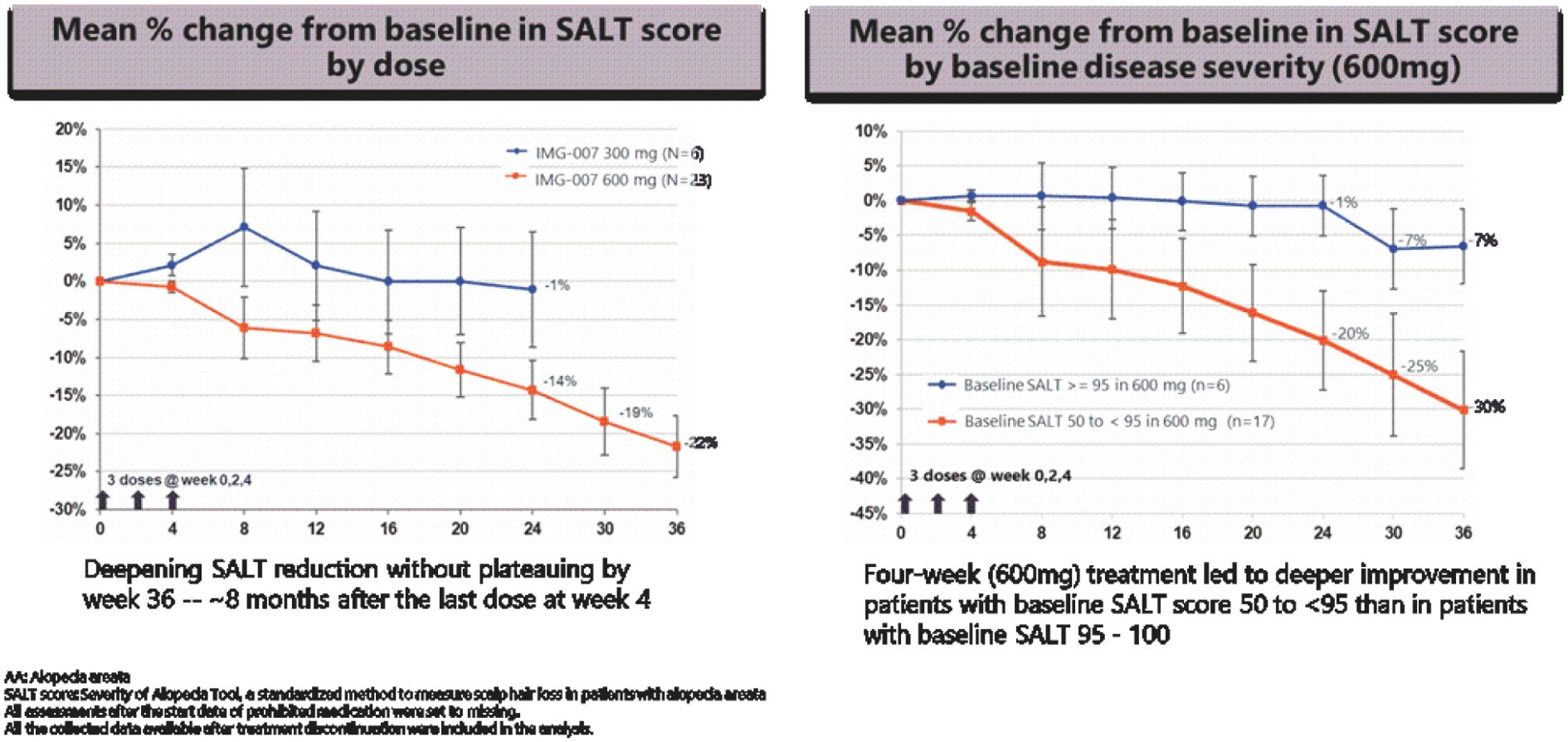
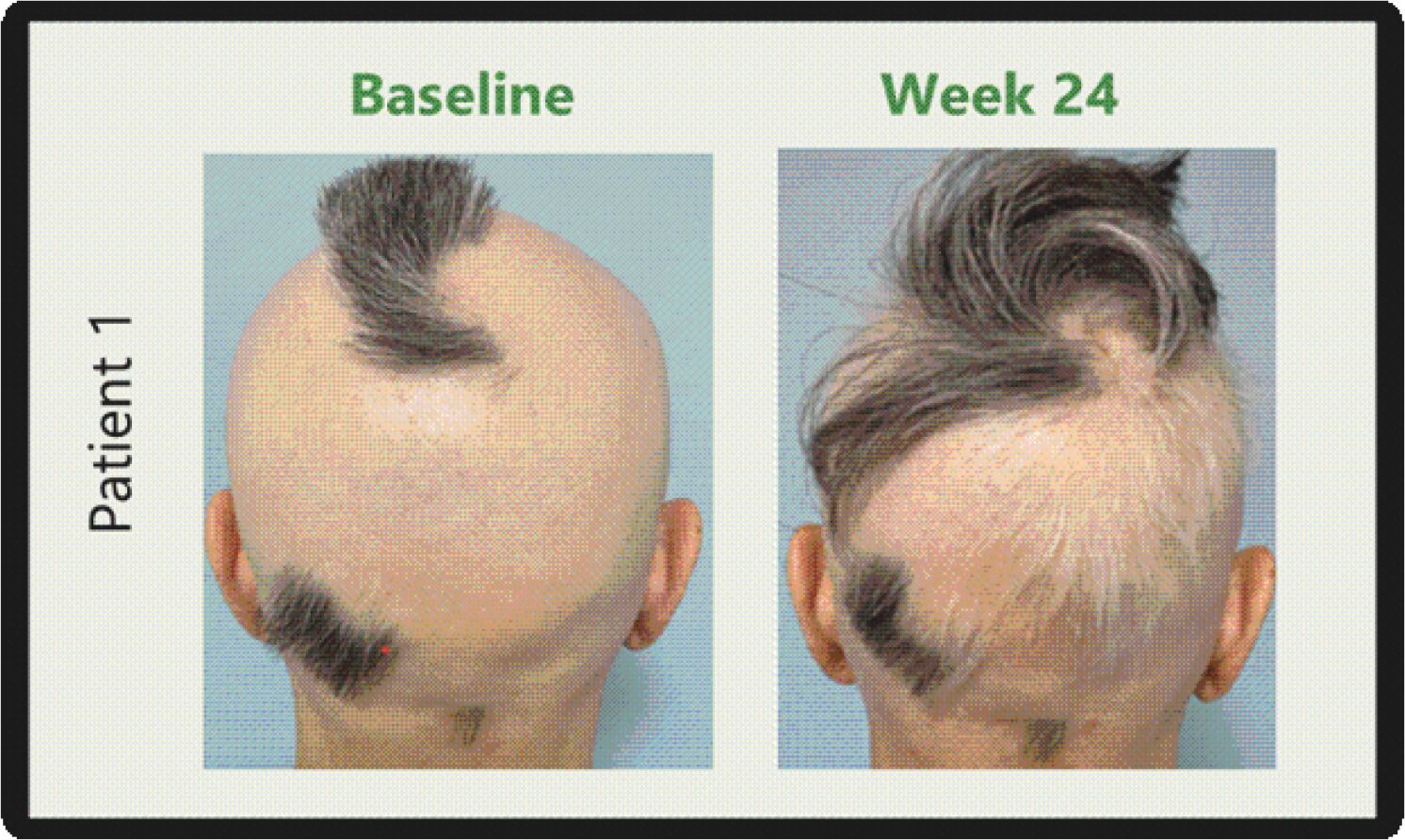
The information in this preliminary prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
Subject to Completion, dated April 2, 2026
PROSPECTUS
2,508,337 Shares of Common Stock

This prospectus relates to the resale by certain of the selling stockholders named in this prospectus (each, a “selling stockholder” and collectively, the “selling stockholders”) of 2,508,337 shares of common stock, par value $0.001 per share, issued in a private placement on July 25, 2025.
We are registering the offer and sale of these shares to satisfy certain registration rights we have granted. We will not receive any of the proceeds from the sale of the shares of common stock by the selling stockholders. We will pay the expenses associated with registering the sales by the selling stockholders, as described in more detail in the section titled “Use of Proceeds” appearing elsewhere in this prospectus.
The selling stockholders may sell the common stock described in this prospectus in a number of different ways and at varying prices. We provide more information about how the selling stockholders may sell their common stock in the section titled “Plan of Distribution” appearing elsewhere in this prospectus.
The selling stockholders may sell any, all or none of the shares and we do not know when or in what amount the selling stockholders may sell their common stock hereunder following the effective date of this registration statement.
Our common stock is listed on The Nasdaq Capital Market (“Nasdaq”) under the symbol “IMA.” On April 1, 2026, the last quoted sale price for our common stock as reported on Nasdaq was $5.05.
We are an “emerging growth company” and a “smaller reporting company” as defined under the federal securities laws, and, as such, may elect to comply with certain reduced public company reporting requirements for future filings.
Investing in our securities involves a high degree of risk. Before making an investment decision, you should review carefully the risks and uncertainties described in the section titled “Risk Factors” beginning on page 6 of this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is , 2026.